Attached files
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
______________
FORM 10-K
______________
FORM 10-K
______________
| (Mark One) | |
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| For the Fiscal Year Ended December 31, 2009 | |
| or | |
| o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from __________ to __________.
Commission File Number: 0-20859
GERON CORPORATION
(Exact name of registrant as specified in its charter)
| Delaware | 75-2287752 |
| (State or other jurisdiction of | (I.R.S. Employer |
| incorporation or organization) | Identification No.) |
| 230 Constitution Drive, Menlo Park, CA | 94025 |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: (650) 473-7700
Securities registered pursuant to Section 12(b) of the
Act:
| Title of each class | Name of each exchange on which registered |
| Common Stock, $0.001 par value | Nasdaq Global Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check
mark if the registrant is a well-known seasoned issuer, as defined in Rule 405
of the Securities Act. Yes o No x
Indicate by check mark if the
registrant is not required to file reports pursuant to Section 13 or Section
15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports
required to be filed by Section 13 or 15(d) of the Securities Exchange Act of
1934 during the preceding 12 months (or for such shorter period that the
registrant was required to file such reports), and (2) has been subject to such
filing requirements for the past 90 days. Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to
Item 405 of Regulation S-K is not contained herein, and will not be contained,
to the best of registrant’s knowledge, in definitive proxy or information
statements incorporated by reference in Part III of this Form 10-K or any
amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated
filer, an accelerated filer, a non-accelerated filer, or a smaller reporting
company. See the definitions of “large accelerated filer”, “accelerated filer”
and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| o | Large accelerated filer | x | Accelerated filer |
| o | Non-accelerated filer (Do not check if a smaller reporting company) | o | Smaller reporting company |
Indicate by check mark whether the
registrant is a shell company (as defined in Rule 12b-2 of the Act).
Yes o No x
The aggregate market value of voting and non-voting common equity held by
non-affiliates of the registrant was approximately $679,017,000 based upon the
closing price of the common stock on June 30, 2009 on the Nasdaq Global Market.
Shares of common stock held by each officer, director and holder of five percent
or more of the outstanding common stock have been excluded in that such persons
may be deemed to be affiliates. This determination of affiliate status is not
necessarily a conclusive determination for other purposes.
As of February 23, 2010, there
were 97,455,463 shares of common stock outstanding.
DOCUMENTS INCORPORATED BY
REFERENCE:
| Form 10-K | |||
| Document | Parts | ||
| Portions of the Registrant’s definitive proxy statement for the 2010 annual meeting of stockholders to be filed pursuant to Regulation 14A within 120 days of the Registrant’s fiscal year ended December 31, 2009 | II, III | ||
Forward-Looking Statements
This annual report on Form 10-K, including “Management’s Discussion and
Analysis of Financial Condition and Results of Operations” in Item 7, contains
forward-looking statements that involve risks and uncertainties, as well as
assumptions that, if they never materialize or prove incorrect, could cause the
results of Geron Corporation (Geron or the Company) to differ materially from
those expressed or implied by such forward-looking statements. All statements
other than statements of historical fact are statements that could be deemed
forward-looking statements. The risks and uncertainties referred to above
include, without limitation, risks inherent in the development and
commercialization of Geron’s potential products, dependence on collaborative
partners, need for additional capital, need for regulatory approvals or
clearances, maintenance of Geron’s intellectual property rights and other risks
that are described herein and that are otherwise described from time to time in
Geron’s Securities and Exchange Commission reports including, but not limited
to, the factors described in Item 1A, “Risk Factors,” of this annual report.
Geron assumes no obligation and does not intend to update these forward-looking
statements.
PART I
ITEM 1. BUSINESS
Overview
Geron is developing first-in-class biopharmaceuticals for the treatment
of cancer and chronic degenerative diseases, including spinal cord injury, heart
failure and diabetes. The company is advancing an anti-cancer drug and a cancer
vaccine that target the enzyme telomerase through multiple clinical trials in
different cancers.
We were incorporated in 1990 under the laws of Delaware. Our principal
executive offices are located at 230 Constitution Drive, Menlo Park, California
94025. Our telephone number is (650) 473-7700.
We make available free of charge on or through our Internet website our
annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on
Form 8-K and all amendments to those reports as soon as reasonably practicable
after they are electronically filed with, or furnished to, the Securities and
Exchange Commission. Our Internet website address is www.geron.com. Information
on our website is not incorporated by reference and does not form a part of this
report. Copies of our annual reports on Form 10-K will be furnished without
charge to any person who submits a written request directed to the attention of
our Secretary, at our offices located at 230 Constitution Drive, Menlo Park,
California 94025.
Major Technology Platforms
Telomeres and Telomerase: Role in Cellular Aging and Cancer
Cells are the building blocks for all tissues in the human body and cell
division plays a critical role in the normal growth, maintenance and repair of
human tissue. However, in the human body, most cell division is a limited
process. Depending on the tissue type, cells generally divide only 60 to 100
times during the course of their normal lifespan.
We and our collaborators have shown that telomeres, located at the ends
of chromosomes, are key genetic elements involved in the regulation of the
cellular aging process. Our work has shown that each time a normal cell divides,
telomeres shorten. Once telomeres reach a certain short length, cell division
halts and the cell enters a state known as replicative senescence or aging.
Thus, this shortening of the telomeres effectively serves as a molecular “clock”
for cellular aging. We and others have shown that when the enzyme telomerase is
introduced into normal cells, it can restore telomere length — reset the “clock”
— thereby increasing the functional lifespan of the cells. Importantly, it does
this without altering the cells’ biology or causing them to become cancerous.
Human telomerase, a complex enzyme, is composed of a ribonucleic acid (RNA)
component, known as hTR, a protein component, known as hTERT, and other
accessory proteins. In 1994, we cloned the gene for hTR, and in 1997, with
collaborators, cloned the gene for hTERT.
2
The 2009 Nobel Prize for Physiology and Medicine was awarded for the
discovery of how chromosomes are protected by telomeres and the enzyme
telomerase. The Nobel laureates were early Geron collaborators, Elizabeth H.
Blackburn and Carol W. Greider, along with Jack W. Szostak.
Our work and that of others has shown that telomerase is not present, or
is present at very low levels, in most normal cells and tissues, but that during
cancer progression, telomerase is abnormally reactivated in all major cancer
types. Our studies have shown that while telomerase does not cause cancer (which
is caused by mutations in oncogenes and tumor suppressor genes), the continued
presence of telomerase enables cancer cells to maintain telomere length,
providing them with indefinite replicative capacity. We and others have shown in
various tumor models that inhibiting telomerase activity results in telomere
shortening and causes aging or death of the cancer cell.
Although telomerase is expressed in nearly all cancer cells, it is not
expressed in most normal cells. That gives telomerase the potential of being
both a universal as well as a highly specific cancer target. This specificity
means that drugs and biologics that attack cancer cells by targeting telomerase
may leave most other cells unaffected, and thus may have fewer side effects than
conventional chemotherapeutic agents that typically affect both cancer and
non-cancer cells.
We are developing anti-cancer therapies based on telomerase inhibitors
and telomerase therapeutic vaccines. Through our licensee, we also intend to
develop products using telomerase as a marker for cancer diagnosis, prognosis,
patient monitoring and screening.
We are also researching compounds that transiently activate telomerase in
senescent cells to restore cell function for the treatment of injuries and
chronic diseases.
Human Embryonic Stem Cells: A Potential Source for the Manufacturing of
Therapeutic Cells
Stem cells generally are self-renewing primitive cells that can develop
into functional, differentiated cells. Human embryonic stem cells (hESCs), which
are derived from very early stage embryos called blastocysts, are unique
because:
- they are pluripotent, which means
they can develop into all cells and tissues in the body, and
- they self-renew indefinitely in the undifferentiated state because they express high levels of telomerase.
The ability of hESCs to divide indefinitely in the undifferentiated state
without losing pluripotency is a unique characteristic that distinguishes them
from all other stem cells discovered to date in humans. We have demonstrated
that hESCs express telomerase continuously, a characteristic of immortal cells.
Other stem cells such as blood or gut stem cells express telomerase at very low
levels or only periodically; they therefore age, limiting their use in research
or therapeutic applications. hESCs can be expanded in culture indefinitely and
hence can be banked for scaled product manufacture.
We intend to use human embryonic stem cell technology to enable the
development of transplantation therapies by providing standard starting material
for the manufacture of therapeutic cells and facilitate pharmaceutical research
and development practices by providing cells for disease models and screening.
Commercial Opportunities for Our Major
Technology Platforms
Oncology
Cancer is a group of diseases characterized by the uncontrolled growth
and spread of abnormal cells. The American Cancer Society estimated that nearly
1.5 million new cancer cases were diagnosed in 2009 and overall annual costs
associated with cancer in 2008 were an estimated $228.1 billion in the United
States alone. Because telomerase is detectable in more than 30 human cancer
types and in the great majority of cancer samples studied, we believe that
telomerase-based drugs could overcome the limitations of current cancer
therapies and potentially be broadly applicable and highly specific drug
treatments for cancer.
3
We and our licensees are developing a range of anti-cancer therapies and
diagnostics, including anti-cancer therapies based on telomerase inhibitors and
telomerase therapeutic vaccines, and diagnostics based on telomerase detection.
We believe telomerase is an ideal target for cancer therapeutics and diagnostics
because it appears to be universal (expressed in all major types of cancers
studied to date), specific (not expressed in most normal cells), and critical
(required for long-term survival of cancer cells). We believe that we have the
dominant patent position in the field of telomerase. Whether it is achieved by
us or our licensees, we believe that progress in the development of
telomerase-based cancer therapeutics and diagnostics will further validate the
importance of telomerase as a cancer target and therefore benefit all of our
telomerase cancer programs.
The following table briefly describes the cancer therapeutic and
diagnostic products being developed by us or our licensees and the stage of
development of these product candidates.
| Patient | ||||||||||
| Product | Development | Enrollment | ||||||||
| Product | Description | Application | Stage | Status | ||||||
| Imetelstat | Telomerase | Chronic Lymphoproliferative | Phase I Trial | Completed | ||||||
| (GRN163L) | Inhibitor | Diseases | (single agent) | |||||||
| Imetelstat | Telomerase | Solid Tumors | Phase I Trial | Open | ||||||
| (GRN163L) | Inhibitor | (single agent) | ||||||||
| Imetelstat | Telomerase | Multiple Myeloma* | Phase I Trial | Completed | ||||||
| (GRN163L) | Inhibitor | (single agent) | ||||||||
| Imetelstat | Telomerase | Non-Small Cell Lung | Phase I Trial | Completed | ||||||
| (GRN163L) | Inhibitor | Cancer* | (combination) | |||||||
| Imetelstat | Telomerase | Breast Cancer* | Phase I/II Trial | Open | ||||||
| (GRN163L) | Inhibitor | (combination) | ||||||||
| Imetelstat | Telomerase | Multiple Myeloma | Phase I Trial | Completed | ||||||
| (GRN163L) | Inhibitor | (combination) | ||||||||
| GRNVAC1 | Telomerase | Acute Myelogenous | Phase II Trial | Completed | ||||||
| Cancer Vaccine | Leukemia (AML) | |||||||||
| * | Initiation of Phase II clinical trials in multiple myeloma, non-small cell lung cancer, breast cancer and essential thrombocythemia is planned for 2010. | |||||||||
| Patient | ||||||||||
| Product | Development | Enrollment | ||||||||
| Licensees | Description | Application | Stage | Status | ||||||
| Merck & Co. | Telomerase | Prostate and Solid Tumors | Phase I Trial | Completed | ||||||
| Cancer Vaccine | ||||||||||
| Sienna Cancer | Telomerase | Bladder Cancer | Preclinical | N/A | ||||||
| Diagnostics | Diagnostic | Development | ||||||||
Telomerase Inhibition (Imetelstat Sodium - GRN163L). Upregulation of telomerase is necessary for
most cancer cells to replicate indefinitely and thereby enable tumor growth and
metastasis. One of our strategies for the development of anti-cancer therapies
is to inhibit telomerase activity in cancer cells. Inhibiting telomerase
activity should result in telomere shortening which can cause aging and death of
cancer cells. Recent data show that telomerase can protect tumor cells from
genomic instability and other forms of cellular stress, suggesting that
inhibiting telomerase can cause a more rapid suppression of tumor growth than
predicted by telomere loss alone. Because telomerase is expressed at very low
levels, if at all, in most normal cells, the telomerase inhibition therapies
described below are being developed with the goal of being less toxic to normal
cells than conventional chemotherapy.
We have designed and synthesized a special class of short-chain nucleic
acid molecules, known as oligonucleotides, which target the template region, or
active site, of telomerase. Our recent work has focused on one of these
oligonucleotides, called imetelstat sodium (originally known as GRN163L). We
have demonstrated that it has highly potent telomerase inhibitory activity at
very low concentrations in biochemical assays, various cellular systems and
animal studies. Imetelstat is a direct enzyme inhibitor,
4
not an antisense
compound. It is smaller (lower molecular weight) than typical antisense
compounds or other oligonucleotide drug candidates and uses a special
thiophosphoramidate chemical backbone, for which we acquired key patents in
March 2002 from Lynx Therapeutics.
Imetelstat sodium (imetelstat) is a 13-mer oligonucleotide N3’-- P5’
thiophosphoramidate (NPS oligonucleotide) that is covalently attached to a C16
(palmitoyl) lipid moiety, which increases potency and improves its
pharmacokinetic and pharmacodynamic properties. Imetelstat binds directly with
high affinity to the template region of the RNA component of human telomerase
(hTR), which lies in the active or catalytic site of hTERT, the telomerase
reverse transcriptase. Imetelstat binding to hTR results in direct, competitive
inhibition of telomerase enzymatic activity.
After completing a series of animal toxicology and preclinical efficacy
studies of imetelstat in 2005 and filing an Investigational New Drug (IND)
application, we received clearance from the U.S. Food and Drug Administration
(FDA) to begin human clinical trials of imetelstat. We sponsored six Phase I or
I/II clinical trials at 22 U.S. medical centers to examine the safety,
tolerability, pharmacokinetics and pharmacodynamics of imetelstat, alone or in
combination with other standard therapies in patients with chronic
lymphoproliferative diseases, solid tumors, multiple myeloma, non-small cell
lung and breast cancer. Four of those trials fulfilled their patient quotas and
completed patient enrollment during the fourth quarter of 2009.
Telomerase inhibition by imetelstat was first demonstrated in humans in
the Phase I single agent trial in patients with relapsed or refractory multiple
myeloma. The early results from this trial were presented at the 2008 American
Society of Hematology annual meeting. Importantly, clinical data from the
ongoing trial showed that imetelstat inhibits telomerase both in the bulk
myeloma fraction as well as the stem cell-containing fraction in patients’ bone
marrow.
Preclinical studies have also demonstrated that imetelstat can inhibit
growth of cancer stem cells from multiple tumor types. These data were presented
during the April 2009 Annual Meeting of the American Association for Cancer
Research. Cancer stem cells capable of clonogenic growth may play an important
role in the regrowth of tumors after initial reduction by standard treatments.
Preclinical study results showed that imetelstat inhibits in vitro cell
colony growth of both primary patient samples and subpopulations from cell lines
containing or enriched for cancer stem cells from myeloma, melanoma, breast,
pancreatic, pediatric glioma, neuroblastoma, prostate, lung and glioblastoma
tumor types. These subpopulations typically show resistance to several
conventional chemotherapeutic agents.
At the November 2009 AACR-NCI-EORTC International Conference on Molecular
Targets and Cancer Therapeutics, we presented interim data on the ongoing trial
of imetelstat in patients with relapsed or refractory solid cancers. These data
showed that with a modified dosing schedule we are achieving exposures to
imetelstat that exceed the levels that have been associated with inhibiting
tumor growth in several models of human cancers.
We have met our main objectives for Phase I of assessing the safety,
tolerability, pharmacokinetics and pharmacodynamics of imetelstat. We have
established our single agent Phase II dose and dosing schedule and are planning
to advance the program to Phase II trials in 2010 in four different
malignancies.
Telomerase Therapeutic Vaccine (GRNVAC1). The goal of therapeutic cancer vaccines is to
“teach” the patient’s own immune system to attack cancer cells while sparing
other cells. This is done by repeatedly exposing the immune system to a
substance (antigen) that is specific to cancer cells in a way that subsequently
induces an immune response to any cells that express that antigen on their
surface. We believe that the characteristics of telomerase make it an ideal
antigen for cancer vaccines.
GRNVAC1 is an autologous product consisting of mature dendritic cells
(the body’s most powerful antigen-presenting cells) pulsed with RNA for the
protein component of human telomerase (hTERT) and a portion of a lysosomal
targeting signal (LAMP). GRNVAC1 is injected into the patient’s skin; from there
the dendritic cells travel to the lymph nodes and instruct cytotoxic T-cells to
kill tumor cells that express telomerase on their surface.
The first clinical study of GRNVAC1 was conducted at Duke University
Medical Center. Data from this Phase I clinical trial in prostate cancer
patients were published in the Journal of Immunology in March 2005. Several small additional Phase I/II trials, which
concluded in 2006, in patients with
5
prostate cancer,
hematologic malignancies and renal cell carcinoma were performed at Duke in
order to optimize the vaccination process. As a result of positive data from
these studies, we brought the vaccine manufacturing process in-house for further
optimization and transferred it to a contract manufacturer.
The Geron-sponsored clinical study of GRNVAC1 is being conducted at six
U.S. medical centers. This Phase II clinical trial is using a prime-boost
vaccination protocol in patients with acute myelogenous leukemia (AML) in
complete clinical remission and examines the safety and feasibility of a
prime-boost vaccination regimen to extend the duration of telomerase immunity.
Also we are evaluating the immune response to GRNVAC1 and exploring the effects
of vaccination on minimal residual disease and relapse rates. This trial
completed patient enrollment in December 2009.
In the AML Phase II trial, patients enter the study in their first or
second complete remission. Prior to or shortly after completing consolidation
chemotherapy, patients undergo leukapheresis to harvest normal peripheral blood
mononuclear cells for vaccine manufacture. Patient mononuclear cells are
differentiated in culture to immature dendritic cells, which are transfected
with messenger RNA encoding hTERT and LAMP. Transfected dendritic cells are
matured, aliquoted and cryopreserved. GRNVAC1 is released for patient dosing
contingent on several product specifications including, identity of mature
dendritic cells, confirmation of positive transfection with hTERT, number of
viable cells per dose after thawing and product sterility. Patients are
vaccinated weekly for six weeks, followed by a rest period of four weeks, and
subsequent boost injections every other week for 12 weeks. Monthly extended
boost injections are then administered until the vaccine product supply is
depleted or the disease relapses.
Twenty patients in the study have received GRNVAC1 product. One patient
relapsed prior to vaccination. GRNVAC1 was found to be safe and generally well
tolerated over multiple vaccinations, including one patient who has received 28
serial vaccinations. Idiopathic thrombocytopenic purpura (grade 4) was reported
in one patient. Other toxicities were mild to moderate, including rash or
headache in 15-20% of patients.
At the December 2009 American Society of Hematology annual meeting we
presented interim data from the Phase II study in patients with AML. At the time
of the presentation, 14 out of 20 patients in the study remained in complete
clinical remission (CR). Median duration of CR, including the patients who had
relapsed, was 12 months. Six of the patients in CR were in the extended boost
phase of vaccination and the duration of their remission since the start of
vaccination ranged from four to 20 months. Four of these six patients are
considered at a high risk of relapse as predicted by their cytogenetics or
because they are in the second CR. Follow-up of the patients for an additional
nine months is required in order to estimate the impact of vaccination on
disease-free survival.
Expression of WT-1, as a marker of minimal residual disease, was
sequentially analyzed by qPCR in 19 patients. The 14 patients who remain in CR
were negative for WT-1, while four of five with clinical relapse were WT-1
positive. One patient was positive for WT-1 prior to vaccination with GRNVAC1
and became WT-1 negative during the course of vaccination.
Patient immune response to telomerase after vaccination with GRNVAC1 was
evaluated using two methods: the delayed-type hypersensitivity (DTH) skin
response and the ELISPOT assay to measure the presence of activated T-cells
specific to hTERT. Positive overall immune responses were detected in 12 out of
20 patients. No correlation has yet emerged between positive immune response and
patient remission status.
In 2004, we acquired rights from Argos Therapeutics, Inc. (formerly
Merix) to commercialize the ex vivo dendritic
cell processing technology used in the Duke clinical trials for telomerase and
other defined tumor-specific antigens. We own the rights to the telomerase
antigen and its use in therapeutic vaccines.
In 2006, we licensed rights from Immunomic Therapeutics, Inc. to the LAMP
antigen targeting sequence for use in cancer vaccines. The LAMP sequence causes
an antigen to which it is attached to be taken up by the lysosomal subcellular
compartment of the cell. This has been shown to increase presentation on MHC
class II molecules, which in turn, can produce greater CD4+ T-cell responses
against the antigen and a more potent and longer lasting overall immune
response.
6
In July 2005, we entered into a worldwide exclusive research, development
and commercialization license agreement with Merck & Co., Inc. for cancer
vaccines targeting telomerase by methods other than dendritic cell delivery. In
December 2007, Merck filed an IND to initiate a clinical trial for their cancer
vaccine candidate that targets telomerase. In 2008, Merck initiated a Phase I
clinical trial of V934/V935, a non-dendritic cell-based cancer vaccine candidate
targeting telomerase. The trial will assess the safety, tolerability and
immunogenicity of the vaccine candidate in patients with solid tumors, including
non-small cell lung cancer and prostate carcinoma. The trial has completed
patient enrollment and boost vaccination and/or follow-up of patients is
ongoing.
Cancer Diagnostics.
Telomerase is a broadly applicable and highly specific marker for cancer because
it has been detected in more than 30 human cancer types and in the great
majority of cancer samples studied. We believe that the detection of telomerase
may have significant clinical utility for cancer diagnosis, prognosis,
monitoring and screening. Current cancer diagnostics apply only to a single or
limited number of cancer types because they rely on molecules expressed only by
particular cancer types. However, telomerase-based diagnostics could potentially
address a broad range of cancers.
We have developed several proprietary assays for the detection of
telomerase which are based on its activity or the presence of its RNA or protein
components. The first-generation assay is the Telomeric Repeat Amplification
Protocol (TRAP) assay which can be used to detect telomerase activity in human
tissue or cells, including clinical samples. The second-generation assays detect
the presence of hTR and hTERT in human tissues and body fluids. We own issued
patents for the detection of telomerase activity and the components of
telomerase, including patents for the TRAP assay and diagnostic methods based on
telomerase detection. Currently, our licensees are selling 11 research-use-only
kits that incorporate our technology.
In 2007, we granted a license to Sienna Cancer Diagnostics (Sienna), an
Australian company, to develop and commercialize methods other than PCR
(polymerase chain reaction) and ELISA (Enzyme-Linked ImmunoSorbent Assay) to
detect telomerase for in vitro cancer
diagnosis. Sienna’s lead product in development is a non-invasive assay that
utilizes Sienna’s proprietary Telomerase Biosensor Technology (TBT) to detect
telomerase activity in urine for the diagnosis of bladder cancer. In
consideration for the license, we received an equity interest in Sienna and are
entitled to receive royalties on future product sales.
Telomerase Activation
We are researching drug candidates to treat various degenerative diseases
by the controlled activation of telomerase. Data published by us and others have
indicated that cellular aging caused by shortening telomeres, which occurs in
numerous tissues throughout the human body, causes or contributes to chronic
degenerative diseases and conditions including bone and marrow diseases,
pulmonary fibrosis, HIV/AIDS, liver disease, macular degeneration,
cardiovascular diseases and impaired wound healing. Controlled activation of
telomerase in normal cells can restore telomere length or slow the rate of loss,
improve functional capacity and increase the proliferative lifespan of cells.
Our approach to the therapeutic use of telomerase activation has included
both small molecule drug discovery and biological methods of restoring
telomerase activity. We have applied proprietary gene transfer technologies,
gene expression systems and small molecule screening technology to discover
therapeutic agents to target, postpone and modulate the destructive genetic
changes that occur in senescent cells.
Our majority-owned subsidiary based in Hong Kong, TA Therapeutics, Ltd.
(TAT), was established to commercially develop products that utilize telomerase
activator drugs to restore the regenerative and functional capacity of cells in
various organ systems that have been impacted by senescence, injury or chronic
disease. TAT is conducting preclinical research with small molecule development
leads. Data from tissue culture studies showed that one such lead compound,
TAT2, significantly activates telomerase and improves replicative capacity and
function, including anti-viral activity in HIV-specific CD8+ T-cells from
HIV/AIDS donors. The data were published in the Journal of Immunology in 2008. We own 75% of TAT and Biotechnology Research Corporation (BRC)
owns 25%.
7
Human Embryonic Stem Cell Therapies
The two properties of hESCs, their immortality and pluripotency, enable
the development of a potential new economic model for cell-based products and
therapeutics, namely the development of products available “on demand.” We have
developed proprietary methods to grow, maintain, and scale the culture of
undifferentiated hESCs that use feeder cell-free and serum-free media with
chemically defined components. Moreover, we have developed scalable processes to
differentiate these cells into therapeutically relevant cells. We have developed
cryopreserved formulations of hESC-derived cells to enable our business model of
delivering “on demand” cells for therapeutic use.
Under our collaboration with Corning Life Sciences, a division of Corning
Incorporated, we are working together to develop synthetic growth surfaces to
replace the biological surface coatings that are widely used today to grow
hESCs. Together our teams have developed a synthetic peptide surface that can be
manufactured into multiple culture vessel formats and directly supports the
growth and differentiation of hESCs. Data on hESC culture and differentiation
were presented by Corning at the 2009 World Stem Cell Summit held in
September.
The following table briefly describes the hESC-derived product candidates
being developed by us or our collaborators and the stage of development of these
product candidates.
| Product | Product Description | Application | Development Stage | |||||
| GRNOPC1 | Oligodendrocytes | Spinal Cord Injury | Phase I Trial * | |||||
| GRNCM1 | Cardiomyocytes | Heart Disease and Screening | Preclinical | |||||
| GRNIC1 | Islets | Type 1 Diabetes | Research | |||||
| GRNCHND1 | Chondrocytes | Osteoarthritis | Research | |||||
| -- | Hepatocytes | ADME Drug Screening | Research | |||||
| GRNVAC2 | Mature Dendritic Cells | Cancer Immunotherapy | Product Research | |||||
| -- | Immature Dendritic Cells | Immune Rejection | Research | |||||
| -- | Osteoblasts | Osteoporosis | Research |
| * | In January 2009, we received clearance from the FDA to begin a clinical trial of GRNOPC1. The trial is currently on clinical hold by the FDA. |
We believe we have a dominant patent position in the field of hESCs. We
own or have licenses to intellectual property covering core inventions and
enabling technologies in this field.
Oligodendrocyte Progenitor Cells for Spinal Cord Injury
(GRNOPC1). The major
neural cells of the central nervous system typically do not regenerate after
injury. If a nerve cell is damaged due to disease or injury, there is no
treatment at present to restore lost function. Patients worldwide suffer from
injury to the nervous system or disorders associated with its degeneration. In
the case of spinal cord injuries, patients are often left partly or wholly
paralyzed because nerve and supporting cells in the spinal cord have been
damaged and cannot regenerate. Such patients are permanently disabled, often
institutionalized and may require life support.
Embryonic stem cell-derived neural cells have been used by researchers to
treat nervous system disorders in animal models. In the case of spinal cord
injuries, neural cells derived from animal embryonic stem cells and injected
into the spinal cord injury site produced significant recovery of the animal’s
ability to move and bear weight.
To apply those observations to humans, we have derived oligodendrocyte
progenitor cells (GRNOPC1) from hESCs. Oligodendrocytes are naturally occurring
cells in the nervous system that have several functions. Oligodendrocytes
produce myelin (insulating layers of cell membrane) that wraps around the axons
of neurons to enable them to conduct electrical impulses. Myelin enables
efficient conduction of nerve impulses in the same manner as insulation prevents
short circuits in an electrical wire. Without myelin, many of the nerves in the
brain and spinal cord cannot function properly. Oligodendrocytes also produce
neurotrophic factors (biologicals that enhance neuronal survival and function)
to support the maintenance of nerve cells. Oligodendrocytes are lost in spinal
cord injury, resulting in myelin and neuronal loss that cause paralysis in many
patients.
8
In our collaboration with researchers at the University of California,
Irvine, we have shown in animal models that GRNOPC1 can improve functional
locomotor behavior after implantation in the injury site seven days after
injury. Histological analysis also provided evidence for the engraftment and
function of these cells. These data were first published in May 2005 in the
Journal of Neuroscience. In additional studies, the lesion site of
animals nine months after injury and subsequent injection of GRNOPC1 was
observed to be essentially filled with GRNOPC1 and myelinated rat axons crossing
the lesion. These animal observations serve as the rationale for the use of
GRNOPC1 in treating spinal cord injuries in humans.
We have developed a functional cryopreserved formulation of GRNOPC1 for
use in clinical trials and have initiated current Good Manufacturing Practices
(cGMP) production of GRNOPC1 in our qualified manufacturing
facilities.
We completed extensive animal toxicology testing that included 24
separate studies in rats and mice requiring more than five billion GRNOPC1
cells. In those preclinical IND-enabling studies, we had observed the occurrence
of occasional epithelial cysts in animals transplanted with GRNOPC1. These cysts
were non-proliferative, confined to the injury site, smaller than the injury
cavity and were not associated with adverse effects on the animals. Data from
these animal and in vivo studies were included in the IND filed with
the FDA.
In January 2009, we received clearance from the FDA to begin the world’s
first human clinical trial of an embryonic stem cell-based therapy using GRNOPC1
for acute spinal cord injury. The study is a Phase I multi-center trial designed
to assess the safety and tolerability of GRNOPC1 in patients with complete ASIA
(American Spinal Injury Association) grade A thoracic spinal cord
injuries.
Since clearance of the IND, Geron has been performing a series of
preclinical studies to expand the clinical program (preclinical expansion
studies) for spinal cord injury beyond patients with complete thoracic injuries.
Our goal is to test the safety and utility of GRNOPC1 in patients with complete
and incomplete (less severe) injuries in both thoracic and cervical
regions.
In one of the preclinical expansion studies, a higher frequency of
animals developed cysts in the injury site than had been seen in numerous
foregoing preclinical studies with clinical grade GRNOPC1, including the
IND-enabling studies. We notified the FDA of the findings from this animal study
and the trial was put on clinical hold in August 2009. As part of ongoing work
to optimize GRNOPC1 manufacturing and product release, we developed new
candidate markers and assays. Data from studies using the new markers were
submitted to the FDA.
Following discussions with the FDA, we have agreed to complete a
confirmatory preclinical study using GRNOPC1 that has been characterized by the
new markers and assays. As part of the ongoing plan to advance clinical
development to cervical patients, we had already initiated that preclinical
study in an animal model of cervical injury. In our discussions, the FDA has
advised us that it concurs with our assessment that positive data from this
study can be used to support both release of the clinical hold related to our
IND and expansion of the clinical trial to cervical patients.
In addition to spinal cord injury, GRNOPC1 may have therapeutic utility
for other central nervous system indications, such as Alzheimer’s disease,
stroke and multiple sclerosis. We have established two separate collaborations
with academic groups to test GRNOPC1 in models of Alzheimer’s disease and
multiple sclerosis.
Cardiomyocytes for Heart Disease (GRNCM1). Heart muscle cells (cardiomyocytes) do not
regenerate during adult life. When heart muscle is damaged by injury or
decreased blood flow, functional contracting heart muscle is replaced with
nonfunctional scar tissue. Congestive heart failure, a common consequence of
heart muscle or valve damage, affects approximately 5.8 million people in the
United States according to the American Heart Association and this year, it is
estimated that about 1.3 million people will have a heart attack, which is the
primary cause of heart muscle damage.
Heart disease can potentially be treated by using cardiomyocytes derived
from hESCs. Researchers have demonstrated proof-of-concept of this approach in
mice. Mouse embryonic stem cells have been used to derive mouse cardiomyocytes.
When injected into the hearts of recipient adult mice, the cardiomyocytes
repopulated the heart tissue and stably integrated into the muscle tissue of the
adult
9
mouse heart. In human
medicine, it is therefore possible that hESC-derived cardiomyocytes could be
developed for cellular transplantation therapy in humans suffering from
congestive heart failure and the damage caused by heart attacks.
We have derived human cardiomyocytes from hESCs (GRNCM1) using a process
that can be scaled for clinical production. GRNCM1 has normal contractile
function and responds appropriately to cardiac drugs. We have transplanted these
cells into animal models of myocardial infarction in which the cells engraft and
improve the left ventricular function compared to those animals receiving
injections without cells. These results were published in Nature Biotechnology in August 2007. We are now conducting preclinical large animal studies
of GRNCM1.
Islet Cells for Diabetes (GRNIC1). According to the Centers for Disease Control,
it is estimated that there are as many as 1.2 million Americans suffering from
Type 1 Diabetes (Insulin Dependent Diabetes Mellitus). Normally, certain cells
in the pancreas, called the islet ß cells, produce insulin which promotes the
uptake of the sugar glucose by cells in the human body. Degeneration of
pancreatic islet ß cells results in a lack of insulin in the bloodstream which
results in diabetes. Although diabetics can be treated with daily injections of
insulin, these injections enable only intermittent glucose control. As a result,
patients with diabetes suffer chronic degeneration of many organs, including the
eye, kidney, nerves and blood vessels. In some cases, patients with diabetes
have received islet ß cells derived from cadavers. However, poor availability of
suitable sources for islet ß cell transplantation make this approach impractical
as a treatment for the growing numbers of individuals suffering from diabetes.
We have derived insulin-producing cells (i.e. similar to pancreatic islet
ß cells) from hESCs. The original derivation method and characterization of our
hESC-derived islets was published in Stem Cells in
August 2007. In 2008, we published data showing the successful engraftment of
these cells in diabetic mice. After transplantation, the engrafted cells
continued to express important pancreatic islet proteins, responded to high
levels of glucose in the blood and extended the survival of recipient
animals.
We have recently modified our differentiation protocols for more robust
generation of pancreatic endoderm and improved yield of islet cells from hESCs.
Preliminary data show that the new protocol generates hESC-derived islet
clusters with improved insulin-producing properties in vitro, and
detailed characterization is underway. We are also testing in vivo
function.
Chondrocytes for Osteoarthritis (GRNCHND1). Osteoarthritis, or Degenerative Joint
Disease, is an extremely common condition characterized by degradation of
cartilage in joints, often accompanied by bone remodeling and bone overgrowth at
the affected joints. According to the Arthritis Foundation, the disease affects
an estimated 27 million adults in the United States, mostly after age 45. The
disease has many causes, but the end result is a structural degradation of joint
cartilage and a failure of chondrocytes (cartilage-forming cells) to repair the
degraded cartilage collagen matrix. Our collaborators have derived chondrocytes
from hESCs. Transplantation studies conducted in rodent models of an articular
cartilage defect show good engraftment and repair of the defect that integrates
well with host cartilage. Our collaborators intend to initiate large animal
studies in 2010.
Dendritic Cells for Cancer Immunotherapy and to Enable Therapeutic Graft
Acceptance. The
hematopoietic system (the circulating cells of blood) is one of the tissues of
the human body that can replenish itself throughout life. One of the cell types
produced by the hematopoietic system is the dendritic cell. Dendritic cells,
depending on their type, can either induce or downmodulate immune responses.
Therefore, dendritic cells derived from hESCs can be used for two purposes: (i)
to upregulate immune responses to particular antigens such as telomerase for
cancer immunotherapy applications; and (ii) to prevent rejection of hESC-derived
therapeutic grafts.
In 2006, we entered into a worldwide exclusive license and collaboration
agreement with the University of Oxford to produce dendritic cells from hESCs.
The scalable production of dendritic cells from hESCs could serve as an
alternative to isolating dendritic cells from each patient, and possibly as a
broadly useful vaccine delivery vehicle. In another form, dendritic cells may
act to block an immune response against an antigen by teaching the immune system
not to attack it – a process known as “tolerizing” the individual to that
antigen. Since the same pluripotent hESC line could be used to generate both
tolerizing dendritic cells and therapeutic cells, co-administration of these two
cell populations could potentially circumvent immune rejection without the need
for immunosuppressive drugs.
10
With our collaborators in Oxford, we have demonstrated that dendritic
cells scalably manufactured from hESCs exhibit the normal functions of naturally
occurring human dendritic cells found in the bloodstream. The data, published in
Regenerative Medicine in July 2009, showed that immature
hESC-derived dendritic cells are able to take up, process and present antigens,
and then, following maturation in the manufacturing process, are able to
migrate, produce pro-inflammatory cytokines and induce specific immune responses
to both tumor and viral antigens in vitro.
Osteoblasts for Osteoporosis and Non-Union Bone Fractures.
Osteoporosis, or loss of
bone density, is a common condition associated with aging and hormonal changes
in post-menopausal women, causing skeletal deformities, back pain and loss of
height. According to the National Osteoporosis Foundation, the disease caused
over 2.0 million fractures in the United States alone in 2005 and these
fractures often occur after minimal trauma and if severe, as in hip fracture,
carry mortality rates as high as 24% for patients age 50 and over. Nearly one in
five hip fracture patients ends up in a nursing home. Total health care costs
for osteoporosis and its complications are estimated at $18 billion per year in
the United States.
The primary cause of the disease is metabolic bone loss (mediated by
osteoclasts–cells which resorb bone) that is incompletely compensated by new
bone formation (mediated by osteoblasts–cells which form new bone). Osteoblast
activity declines over the human lifespan and fails to keep pace with the
increasing activity of osteoclasts, resulting in progressive loss of bone
density leading to fracture, pain and deformity.
Our collaborators have made osteoblasts from hESCs and conducted early
tests in animals. Continued development of this cell type includes further
testing to confirm cell engraftment and enhancement of derivation protocols to
improve production yields.
Products for Research and Development
Immortalized Cells for Research. Scientists study specific cells from targeted
tissues in order to understand their biological function. For these studies,
cells are usually isolated from tissue and maintained in culture. The
progressive changes in biological activity, morphology and proliferation as a
result of normal cell aging in tissue culture potentially limit the utility of
these cells in serial experiments and long-term research. Because of these
limitations, most research laboratories utilize transformed cell lines for their
studies. Cells can be transformed by using viruses which ultimately cause the
cells to grow indefinitely in culture. However, such immortalized cell lines
have abnormal characteristics compared to non-transformed cells. For this
reason, they are not good models of normal tissue in the human body.
Telomerase-immortalized cells may be ideal for use in biological research
because these cells proliferate indefinitely and function in culture in the same
manner as the normal, mortal cells from which they were derived. Moreover,
telomerase-immortalized cells can function in the body to form normal tissue and
their capacity to differentiate into mature tissue is maintained. The ability of
these cells to maintain normal physical and biological characteristics while
retaining proliferative capacity allows them to be a constant source of cells
for repeat and long-term studies of the function of cells both in culture and in
the body. Telomerase-immortalized cells can be used to study any of the normal
biological pathways in cells and can be used to screen for factors which
influence the appropriate function of those cells. Moreover, cells taken from
diseased tissues which are then telomerase-immortalized in culture can be used
to explore the mechanism of the disease process and to develop interventions to
prevent or treat that disease.
Through our licensees, we make telomerase-immortalized cell lines
commercially available to the research market and to companies for basic
research and for use in drug discovery and biologics production applications. We
have granted royalty-bearing licenses to the American Type Culture Collection
and Lonza Walkersville, Inc. (formerly Cambrex BioSciences) under which these
organizations produce and sell telomerase-immortalized cells for both academic
research and commercial drug discovery. We have also licensed the telomerase
gene to a number of pharmaceutical and biotechnology companies for use in their
internal research programs.
hESC-Derived Cells for Drug
Discovery, Development and Toxicology. Three of the major hurdles of pharmaceutical
drug development are: (i) identifying compounds with activity in diseased
tissue; (ii) understanding the metabolism and biodistribution of the compound;
and (iii) determining the potential
11
toxic side effects of
the compound. Undesirable activity of a compound being evaluated as a drug
candidate in any one of these areas can impact the development and
commercialization of the drug. The earlier in development that a compound is
found to have undesirable characteristics, the faster these characteristics can
be potentially corrected. This potentially translates into reduced costs and
time in drug development, and less harmful patient exposure in clinical trials.
While Geron’s focus is on the development of hESC-derived cells for
therapeutic application, each of the cell types we are developing could also be
useful in drug discovery applications. In June 2009, we entered into a global
exclusive license and alliance agreement with GE Healthcare UK Limited (GE
Healthcare) to develop and commercialize cellular assay products derived from
hESCs for use in drug discovery, development and toxicity
screening.
Two particularly important cell types for use in in vitro cell-based
assays for metabolism studies and toxicity screening are hepatocytes (liver
cells) and cardiomyocytes (heart muscle cells).
Hepatocytes are responsible for metabolizing most compounds and can
therefore be used to predict how drugs are metabolized, how they might interact
with each other in the body, and to what extent they may be toxic to the liver.
Another key step in drug development is to understand whether or not a drug will
interrupt the normal function of cardiomyocytes in the heart. Cardiotoxicity and
hepatotoxicity are the principal reasons clinical trials have been halted and
approved drugs have been withdrawn from market.
Currently, animal models, primary human tissue and cell lines are used to
assess drug metabolism and toxicities. However, these systems have certain
limitations. Animal models have an important role in drug metabolism and
toxicity studies, but they are not fully reliable predictors of human responses
because of basic physiological differences between species. It is not uncommon
for the development of a drug to be halted during clinical trials because animal
systems did not predict the drug’s metabolism or toxicity in humans. A human
in vitro system commonly used is fresh primary human
liver tissue and cells, but access is very limited and the tissue can be
variable depending on the donor or the methods used in processing or culturing
the samples. Transformed human cell lines have been generated to address supply
or variability, but the lines available today do not have the same attributes as
their normal counterparts in the body. For example, human hepatocytes must be
transformed (genetically altered) in order to maintain their proliferative
capacity in culture, and cell lines used in cardiotoxicity studies are often
non-cardiac cells modified to express particular ion channels and thus do not
reflect the normal physiology of a cardiomyocyte.
In contrast, fully functional cells manufactured in bulk from hESCs could
be a reliable, uniform and predictive new tool for pharmaceutical companies to
perform in vitro metabolism, biodistribution, drug-drug
interaction and toxicity testing of drug development candidates.
There is active interest in the development of predictive, robust and
cost-effective in vitro assays from stem cells for drug research and
development and it is expected that the pharmaceutical industry will embrace
cell-based assays derived from hESCs when they become available. It has been
reported that clinical safety and toxicology account for approximately 30% of
drug attrition in the clinic. It is also important to highlight the health
impact and possible risk to patients when toxicities are not detected early in
development. The FDA has called for new and more predictive tools for early drug
safety studies and may encourage widespread adoption of effective hESC-derived
cellular assay products.
The first product being developed under the Geron-GE Healthcare alliance
is human cardiomyocytes derived from hESCs. hESC-derived cardiomyocytes exhibit
the normal electrophysiological function of human ventricular myocytes and
respond appropriately when exposed to cardiac drugs, including drugs that block
hERG channels. Robust sodium and calcium currents with the expected
pharmacological responses are present and well-suited for screening assays.
hESC-derived cardiomyocytes could, for the first time, allow the prediction of
drug effects on the human heart.
Geron has been working with ChanTest, a leading provider of ion channel
screening services, to confirm that hESC-derived cardiomyocytes display
electrophysiological properties of normal human cardiomyocytes and contain the
key voltage-gated ion channels operating in a normal cellular background.
12
Another cellular assay product to be developed under the Geron-GE
Healthcare alliance is human hepatocytes derived from hESCs. hESC-derived
hepatocytes show a number of metabolic functions of human hepatocytes, including
expression of members of the cytochrome P450 family of enzymes, which are
responsible for drug metabolism.
Nuclear Transfer: Agriculture/Biologics
Nuclear transfer is a method for producing animals (clones) whose nuclear
genetic material is derived solely from a donor cell from an individual animal.
In this process, the nucleus containing the chromosomal DNA is removed from the
animal egg cell and subsequently replaced with a nucleus from a donor somatic
(non-reproductive) cell. Fusion between the resulting egg cell and the donor
somatic nucleus results in a new cell which gains a complete set of chromosomes
derived entirely from the donor nucleus. Mitochondrial DNA, providing some of
the genes for energy production, resides outside the nucleus and is provided by
the egg. After a brief culture period that enables the reconstituted egg cell to
initiate embryonic development, the early embryo is implanted into the uterus of
a female animal, where it can fully develop and result in the live birth of a
cloned offspring animal. The offspring is essentially a genetic clone of
(genetically identical to) the animal from which the donor nucleus was obtained.
In early 1997, Dr. Ian Wilmut and his colleagues at the Roslin Institute
were the first to demonstrate, with the birth of Dolly the sheep, that the
nucleus of an adult cell can be transferred to an enucleated egg to create
cloned offspring. The birth of Dolly was significant because it demonstrated the
ability of egg cell cytoplasm, the portion of the egg outside of the nucleus, to
reprogram an adult somatic nucleus. Reprogramming enables the adult somatic cell
nucleus to express all the genes required for the full embryonic development of
the animal. In addition to sheep, the technique has been used to clone mice,
rats, goats, cattle, rabbits, cats, dogs and pigs from donor cells and
enucleated eggs from each respective animal species. In 1999, we acquired Roslin
Bio-Med Ltd., a commercial subsidiary of the Roslin Institute, and an exclusive
license to the use of nuclear transfer technology for multiple applications in
animal and human biology.
Agriculture. Our
nuclear transfer technology can be used for applications in agriculture that
could improve livestock by producing unlimited numbers of genetically identical
animals with superior commercial qualities. Such applications can be extended to
major agricultural sectors, such as beef, dairy, pork and poultry, to provide
large numbers of animals with superior characteristics of disease resistance,
longevity, growth rate or product quality. In January 2008, the FDA issued its
final risk assessment concluding that meat and milk from healthy cloned animals
and their offspring are as safe as those from ordinary animals, effectively
removing the last U.S. regulatory barrier to the marketing of meat and milk from
cloned cattle, pigs and goats.
Transgenic Animals.
Our nuclear transfer technology can be applied to clone animals that have been
genetically engineered to produce proteins for human therapeutic or industrial
use. For example, herds which carry the genes to make human antibodies could be
cloned, thereby allowing for the large-scale production of therapeutic
antibodies or vaccines.
In previous years, we granted a number of licenses to our nuclear
transfer technology to companies who are utilizing it for applications in
agriculture and production of biologicals. In 2005, following successes in three
patent interference proceedings, we formed a joint venture company, Start
Licensing, Inc. (Start), with Exeter Life Sciences, Inc. (Exeter). In August
2008, Start merged with ViaGen, Inc. (ViaGen), a subsidiary of Exeter. The
merger of Start and ViaGen combined the full breadth of intellectual property
rights to nuclear transfer cloning technology, including that developed at the
Roslin Institute for cloning Dolly the sheep, with in-house state-of-the-art
breeding services and expertise in advanced reproductive technologies,
particularly in cloning animals, to provide a one-stop licensing and operating
company. We have retained all rights for use of nuclear transfer technology in
human cells. We own a 28% equity interest in ViaGen.
13
Patents and Proprietary
Technology
A broad intellectual property portfolio of issued patents and pending
patent applications supports our internal product development, collaborations
and licensing relationships. It is also the asset on which our out-licensing
activities are based. As of December 31, 2009, we own or have licensed 184
issued or allowed United States patents, 367 granted or accepted foreign patents
and 341 patent applications that are pending around the world.
Our policy is to seek appropriate patent protection for inventions in our
principal technology platforms — telomerase and human embryonic stem cells — as
well as ancillary technologies that support these platforms or otherwise provide
a competitive advantage or commercial benefit to us. We achieve this by filing
patent applications for discoveries made by our scientists, as well as those
that we make in conjunction with our scientific collaborators and strategic
partners. Typically, although not always, we file patent applications in the
United States and in major markets around the world. We determine the
jurisdictions in which to file a particular patent family based on factors
including: the technology involved; projected market opportunity for our
products; and other relevant commercial activities in the jurisdiction in
question. A typical patent application family includes applications in the
United States, Canada, Europe, Japan, China, India, Australia and South Korea.
In addition, where appropriate, we obtain licenses from other organizations to
patent filings that may be useful in advancing our scientific and product
development programs or proprietary position in a technology. The jurisdictions
in which those in-licensed patents are filed may have already been selected by
the licensing organization from which we obtain the rights; often they are filed
in fewer countries than our internal filings.
The development of biotechnology products, including ours, typically
includes the early development of a platform technology base, followed by rounds
of increasingly focused innovation around a product opportunity, including
identification and definition of a specific product candidate, manufacturing
processes, product formulation and administration methods. The result of this is
that biotechnology products are often protected by several families of patent
filings that are filed at different times during product development and cover
different aspects of the product. Consequently, earlier filed, broad technology
platform patents will usually expire ahead of patents covering later
developments such as product formulations, so that patent expirations on a
product may span several years. Patent coverage may also vary from country to
country based on the scope of available patent protection. There are also
limited opportunities to obtain extension of patent coverage for a product in
certain countries, which add further complexity to the determination of patent
life.
With the foregoing in mind, below we provide an overview of the patent
protection for our major programs. It is important to note that all of our
product candidates are still under development, so further innovation and
associated patent filings may provide additional patent coverage. Furthermore,
the patent expiration ranges given are for the U.S. only and only for patent
families where patents have already issued (i.e., they do not account for
recently filed patent families where the expiration date of patents yet to issue
may be after the last expiration date given here). The stated expiration dates
also do not account for potential patent extensions that may be available. The
information provided here should be reviewed in the context of the section
entitled “Risks Related to Protecting Our Intellectual Property” that begins on
page 24.
We endeavor to monitor worldwide
patent filings by third parties that are relevant to our business. Based on this
monitoring, we may determine that an action is appropriate to protect our
business interests. Such actions may include negotiating patent licenses where
appropriate, filing oppositions or reexaminations against a patent, or filing a
request for the declaration of an interference with a U.S. patent application or
issued patent. Similarly, third parties may take similar actions against our
patents. By way of example, in 2005 we were involved in interference proceedings
that we had initiated at the U.S. Patent and Trademark Office involving patents
and patent applications for nuclear transfer technology; judgments in those
actions were entered in our favor. In 2009, we initiated a patent interference
proceeding involving patent rights relating to the production of endoderm cells
from hESCs, and that proceeding is currently ongoing. We are currently also
involved in patent opposition proceedings before the European Patent Office and
the Australian Patent Office both as the party holding the opposed patent, and
in opposition to patents granted or proposed to be granted to another
entity.
14
Telomerase
Our telomerase platform is the mainstay of our oncology program and it
serves as the basis for other product opportunities. The table on page 4
describes the telomerase products currently in development by Geron and our
partners. As of December 31, 2009, our patent portfolio includes 124 issued or
allowed U.S. patents, 216 granted or accepted foreign patents and 114 patent
applications pending worldwide relating to our telomerase-based product
opportunities. These include patents covering the cloned genes that encode the
RNA component (hTR) and the catalytic protein component (hTERT) of human
telomerase. Related issued and pending patents cover cells that are immortalized
by expression of recombinant hTERT, cancer diagnostics based on detecting the
expression of telomerase in cancer cells, and telomerase inhibitors for use as
cancer therapeutics. We also own patents covering novel amidate oligonucleotide
chemistry that we employ in our telomerase inhibitor program, and methods of
manufacturing these oligonucleotides.
Imetelstat (GRN163L): Our patent rights relevant to imetelstat include those covering the
nucleic acid sequence of hTR on which imetelstat is based; amidate
oligonucleotide chemistry employed in imetelstat; and manufacturing processes
for the drug. These patents are wholly owned by Geron. The expiration dates on
these patents range from 2014 to 2025.
GRNVAC1: Our patent
rights relevant to GRNVAC1 include those covering hTERT; RNA-pulsed dendritic
cells; and the LAMP sequence. We co-own the hTERT patents with the University of
Colorado and hold an exclusive license to the Colorado rights. The patents
covering the RNA-pulsed dendritic cell technology are owned by Duke University,
and we hold co-exclusive license right to those patents from Argos, Inc. The
patents covering the LAMP sequence are owned by Johns Hopkins University and we
hold an exclusive license under those patents to use the LAMP sequence in
conjunction with telomerase from Immunomic Therapeutics, Inc. The expiration
dates on these patents range from 2014 to 2020.
Merck V934 / V935:
We have licensed our hTERT patent rights to Merck for the development of this
product. The relevant issued patents will expire in 2016.
Human Embryonic Stem Cells
Our human embryonic stem cell (hESC) platform serves as the basis for the
development of product candidates that are listed in the table on page 8. As of
December 31, 2009, our patent portfolio includes 39 issued or allowed United
States patents, 100 granted or accepted foreign patents and 213 patent
applications pending worldwide relating to our hESC-based product opportunities.
This portfolio includes: foundational hESC patents licensed to us (exclusively
and non-exclusively, varying by field of use) from the University of
Wisconsin-Madison, or WARF; and patent families exclusively licensed to us by
the University of California, the University of Oxford and the Robarts Research
Institute of the University of Western Ontario. However, the majority of patent
filings in this portfolio is owned by Geron and covers technologies that we have
developed to enable scalable manufacturing of various cell types from
hESCs.
By way of example, our hESC portfolio includes patents and patent
applications covering technologies that we believe will facilitate the
commercial-scale production of hESCs, such as methods for growing the cells
without the need for cell feeder layers, and novel synthetic growth surfaces
that we are developing in conjunction with Corning Life Sciences. We also own or
have licensed patent rights covering cell types that can be made from hESCs,
including hepatocytes (liver cells), cardiomyocytes (heart muscle cells), neural
cells (nerve cells, including dopaminergic neurons and oligodendrocytes),
chondrocytes (cartilage cells), pancreatic islet ß cells, osteoblasts (bone
cells), hematopoietic cells (blood-forming cells) and dendritic cells.
GRNOPC1: Our patent
rights relevant to GRNOPC1 include rights licensed from WARF and the University
of California (both licensed exclusively for this product candidate), and
various Geron-owned patent families covering the growth of hESCs and their
differentiation into neural cells. The expiration dates on these patents range
from 2015 to 2024.
GRNCM1: Our patent
rights relevant to GRNCM1 include rights licensed from WARF (licensed
exclusively for this product candidate), and various Geron-owned patent families
covering the growth of hESCs and their differentiation into cardiomyocytes. The
expiration dates on these patents range from 2015 to 2025.
15
GRNIC1: Our patent
rights relevant to GRNIC1 include rights licensed from WARF (licensed
exclusively for this product candidate), and various Geron-owned patent families
covering the growth of hESCs and their differentiation into pancreatic islet
cells. The expiration dates on these patents range from 2015 to
2023.
GRNVAC2: Our patent
rights relevant to GRNVAC2 include rights licensed from WARF (licensed
non-exclusively for this product candidate) and the University of Oxford and the
Robarts Research Institute of the University of Western Ontario (both licensed
exclusively for this product candidate), and various Geron-owned patent families
covering the growth of hESCs and their differentiation into dendritic cells. The
expiration dates on these patents range from 2015 to 2022.
GE Healthcare Product Candidates (Various): Our alliance agreement with GE Healthcare
includes a license grant from Geron to GE Healthcare under Geron’s hESC patents
to commercialize any hESC-derived cell type for drug discovery applications. GE
Healthcare plans to focus its initial product development efforts on hepatocytes
and cardiomyocytes. Our patents on these particular cell types expire in the
ranges of 2020 to 2023 and 2025 to 2026, respectively.
Nuclear Transfer
ViaGen, Inc.: A
third technology platform, nuclear transfer, is protected by the patent rights
that we purchased in 1999 with the acquisition of the U.K. company Roslin
Bio-Med, which we now operate as Geron Bio-Med. We license these rights to
ViaGen, Inc., in which we hold a 28% equity stake, for use in non-human animal
cloning applications. As of December 31, 2009, 21 United States patents have now
been issued or been allowed, and 51 foreign patents have been granted or
accepted. In addition, we have 14 pending patent applications worldwide relating
to nuclear transfer. The expiration dates of these patents are in
2016.
Government Regulation
Regulation by governmental authorities in the United States and other
countries is a significant factor in the development, manufacture and marketing
of our proposed products and in our ongoing research and product development
activities. The nature and extent to which such regulation applies to us will
vary depending on the nature of any products which may be developed by us. We
anticipate that many, if not all, of our proposed products will require
regulatory approval by governmental agencies prior to commercialization. In
particular, human therapeutic products are subject to rigorous preclinical and
clinical testing and other approval procedures of the FDA and similar regulatory
authorities in European and other countries. Various governmental statutes and
regulations also govern or influence testing, manufacturing, safety, labeling,
storage and recordkeeping related to such products and their marketing. The
process of obtaining these approvals and the subsequent compliance with
appropriate statutes and regulations require the expenditure of substantial time
and money, and there can be no guarantee that approvals will be
granted.
FDA Approval Process
Prior to commencement of clinical studies involving humans, preclinical
testing of new pharmaceutical products is generally conducted on animals in the
laboratory to evaluate the potential efficacy and safety of the product
candidate. The results of these studies are submitted to the FDA as part of an
IND application, which must become effective before clinical testing in humans
can begin. Typically, human clinical evaluation involves a time-consuming and
costly three-phase process. In Phase I, clinical trials are conducted with a
small number of people to assess safety and to evaluate the pattern of drug
distribution and metabolism within the body. In Phase II, clinical trials are
conducted with groups of patients afflicted with a specific disease in order to
determine preliminary efficacy, optimal dosages and expanded evidence of safety.
(In some cases, an initial trial is conducted in diseased patients to assess
both preliminary efficacy and preliminary safety and patterns of drug metabolism
and distribution, in which case it is referred to as a Phase I/II trial.) In
Phase III, large-scale, multi-center, comparative trials are conducted with
patients afflicted with a target disease in order to provide enough data to
demonstrate the efficacy and safety required by the FDA. The FDA closely
monitors the progress of each of the three phases of clinical testing and may,
at its discretion, re-evaluate, alter, suspend, or terminate the testing
16
based upon the data
which have been accumulated to that point and its assessment of the risk/benefit
ratio to the patient. All adverse events must be reported to the FDA. Monitoring
of all aspects of the study to minimize risks is a continuing process.
The results of the preclinical and clinical testing on non-biologic drugs
and certain diagnostic drugs are submitted to the FDA in the form of a New Drug
Application (NDA) for approval prior to commencement of commercial sales. In the
case of vaccines or gene and cell therapies, the results of clinical trials are
submitted as a Biologics License Application (BLA). In responding to an NDA/BLA
submission, the FDA may grant marketing approval, may request additional
information, may deny the application if it determines that the application does
not provide an adequate basis for approval, and may also refuse to review an
application that has been submitted if it determines that the application does
not provide an adequate basis for filing and review. There can be no assurance
that approvals will be granted on a timely basis, if at all, for any of our
proposed products.
European and Other Regulatory Approval
Whether or not FDA approval has been obtained, approval of a product by
comparable regulatory authorities in Europe and other countries will be
necessary prior to commencement of marketing the product in such countries. The
regulatory authorities in each country may impose their own requirements and may
refuse to grant an approval, or may require additional data before granting it,
even though the relevant product has been approved by the FDA or another
authority. As with the FDA, the regulatory authorities in the European Union
(EU) and other developed countries have lengthy approval processes for
pharmaceutical products. The process for gaining approval in particular
countries varies, but generally follows a similar sequence to that described for
FDA approval. In Europe, the European Committee for Proprietary Medicinal
Products provides a mechanism for EU-member states to exchange information on
all aspects of product licensing. The EU has established a European agency for
the evaluation of medical products, with both a centralized community procedure
and a decentralized procedure, the latter being based on the principle of
licensing within one member country followed by mutual recognition by the other
member countries.
Other Regulations
We are also subject to various U.S. federal, state, local and
international laws, regulations and recommendations relating to safe working
conditions, laboratory and manufacturing practices and the use and disposal of
hazardous or potentially hazardous substances, including radioactive compounds
and infectious disease agents, used in connection with our business. We cannot
accurately predict the extent of government regulation which might result from
future legislation or administrative action.
Scientific Consultants
We have consulting agreements with a number of leading academic
scientists and clinicians. These individuals serve as key consultants or as
members of “clinical focus group panels” with respect to our product development
programs and strategies. We use consultants to provide us with expert advice and
consultation on our scientific programs and strategies, as well as on the
ethical aspects of our work. They also serve as important contacts for us
throughout the broader scientific community. They are distinguished scientists
and clinicians with expertise in numerous scientific and medical fields,
including embryonic stem cells, nuclear transfer and telomere and telomerase
biology, developmental biology, cellular biology, molecular biology, oncology,
spinal cord injury, heart disease and diabetes.
We retain each consultant according to the terms of a consulting
agreement. Under such agreements, we pay them a consulting fee and reimburse
them for out-of-pocket expenses incurred in performing their services for us. In
addition, some consultants hold options to purchase our common stock and
restricted stock awards, subject to the vesting requirements contained in the
consulting agreements. Our consultants may be employed by institutions other
than ours, and therefore may have commitments to, or consulting or advisory
agreements with, other entities or academic institutions that may limit their
availability to us.
17
Executive Officers of the
Company
The following table sets forth
certain information with respect to our executive officers:
| Name | Age | Position | ||
| Thomas B. Okarma, Ph.D., M.D. | 64 | President, Chief Executive Officer and | ||
| Director | ||||
| David L. Greenwood | 58 | Executive Vice President, Chief Financial | ||
| Officer, Treasurer and Secretary | ||||
| Stephen M. Kelsey, M.D., F.R.C.P., F.R.C.Path. | 49 | Executive Vice President, Chief Medical | ||
| Officer, Oncology | ||||
| David J. Earp, J.D., Ph.D. | 45 | Senior Vice President, Business Development | ||
| and Chief Patent Counsel | ||||
| Melissa A. Kelly Behrs | 46 | Senior Vice President, Therapeutic | ||
| Development, Oncology | ||||
| Jane S. Lebkowski, Ph.D. | 54 | Senior Vice President, Chief Scientific Officer, | ||
| Regenerative Medicine | ||||
| Katharine E. Spink, Ph.D. | 35 | Vice President Operations, Regenerative | ||
| Medicine Programs |
Thomas B. Okarma, Ph.D., M.D., has served as our President, Chief Executive
Officer and a member of our Board of Directors since July 1999. He is also a
director of Geron Bio-Med Limited, a United Kingdom company and Geron’s
wholly-owned subsidiary, and TA Therapeutics, Ltd., a Hong Kong company and
Geron’s majority-owned subsidiary. From May 1998 until July 1999, Dr. Okarma was
Vice President of Research and Development. From December 1997 until May 1998,
Dr. Okarma was Vice President of Cell Therapies. Dr. Okarma currently serves on
the Board of BIO and was Chairman of the Board of Overseers of Dartmouth Medical
School from 2000 to 2007. In 1985, Dr. Okarma founded Applied Immune Sciences,
Inc. and served initially as Vice President of Research and Development and then
as chairman, chief executive officer and a director of Applied Immune Sciences,
until 1995 when it was acquired by Rhone-Poulenc Rorer. Dr. Okarma was a Senior
Vice President at Rhone-Poulenc Rorer from the time of the acquisition of
Applied Immune Sciences until December 1996. From 1980 to 1992, Dr. Okarma was a
member of the faculty of the Department of Medicine at Stanford University
School of Medicine. Dr. Okarma holds a A.B. from Dartmouth College, a M.D. and
Ph.D. from Stanford University and an executive M.B.A. from Stanford Graduate
School of Business.
David L. Greenwood has served as our Chief Financial Officer, Treasurer and Secretary since
August 1995 and our Executive Vice President since January 2004. He is also a
director of our wholly-owned subsidiary, Geron Bio-Med Limited, our
majority-owned subsidiary, TA Therapeutics, Ltd., ViaGen, Inc., an Arizona
corporation, and Clone International, an Australian company. From August 1999
until January 2004, Mr. Greenwood also served as our Senior Vice President of
Corporate Development. From April 1997 until August 1999, Mr. Greenwood served
as our Vice President of Corporate Development. He also serves on the Board of
Regents for Pacific Lutheran University. From 1979 until joining us, Mr.
Greenwood held various positions with J.P. Morgan & Co. Incorporated, an
international banking firm. Mr. Greenwood holds a B.A. from Pacific Lutheran
University and a M.B.A. from Harvard Business School.
Stephen M. Kelsey, M.D., F.R.C.P., F.R.C.Path., has served as our as Executive Vice President
and Chief Medical Officer, Oncology since April 2009. From June 2002 until April
2009, Dr. Kelsey held various positions at Genentech, Inc., most recently as
vice president, clinical hematology/oncology. From June 2000 to June 2002, Dr.
Kelsey was the director of clinical affairs at Pharmacia Corporation (SUGEN,
Inc.) in South San Francisco and director of global clinical development
(oncology) at Pharmacia Corporation in Milan, Italy. From July 1993 to June
2000, Dr. Kelsey served as a senior lecturer in hematology/oncology at St.
Bartholomews and the Royal London School of Medicine and Dentistry and visiting
fellow at Vancouver General Hospital and Terry Fox Laboratories. Dr. Kelsey
earned his B.Sc. in Pharmacology, M.B., Ch.B., and Doctorate of Medicine (M.D.)
degrees from the University of Birmingham in the United Kingdom.
18
David J. Earp, J.D., Ph.D., has served as our Senior Vice President of Business Development and
Chief Patent Counsel since May 2004. He is also a director of our majority-owned
subsidiary, TA Therapeutics, Ltd. and ViaGen, Inc., an Arizona corporation. From
October 1999 until May 2004, Dr. Earp served as our Vice President of
Intellectual Property. From 1992 until joining us in June 1999, Dr. Earp was
with the intellectual property law firm of Klarquist Sparkman, LLP. Dr. Earp
holds a B.Sc. in microbiology from the University of Leeds, England, a Ph.D.
from the biochemistry department of The University of Cambridge, England, and
conducted postdoctoral research at the University of California at
Berkeley/U.S.D.A. Plant Gene Expression Center. He received his J.D. from the
Northwestern School of Law of Lewis and Clark College in Portland,
Oregon.
Melissa A. Kelly Behrs has served as our Senior Vice President, Therapeutic Development,
Oncology since January 2007. Ms. Behrs served as our Vice President of Oncology
from January 2003 until January 2007. From April 2002 until January 2003, Ms.
Behrs served as our Vice President of Corporate Development. From April 2001
until April 2002, Ms. Behrs served as our General Manager of Research and
Development Technologies. Ms. Behrs joined us in November 1998 as Director of
Corporate Development. From 1990 to 1998, Ms. Behrs worked at Genetics
Institute, Inc., serving initially as assistant treasurer and then as associate
director of preclinical operations where she was responsible for all business
development, regulatory, and project management activities for the preclinical
development function. Ms. Behrs received a B.S. from Boston College and an
M.B.A. from Babson College.
Jane S. Lebkowski, Ph.D., has served as our Senior Vice President, Chief Scientific Officer,
Regenerative Medicine since February 2009 and Senior Vice President of
Regenerative Medicine since January 2004. From August 1999 until January 2004,
Dr. Lebkowski served as our Vice President of Regenerative Medicine. From April
1998 until August 1999, Dr. Lebkowski served as our Senior Director, Cell and
Gene Therapies. From 1986 until joining us in 1998, Dr. Lebkowski served as vice
president, research and development at Applied Immune Sciences. In 1995, Applied
Immune Sciences was acquired by Rhone-Poulenc Rorer, at which time Dr. Lebkowski
was appointed vice president, discovery & product development. Dr. Lebkowski
received a B.S. in chemistry and biology from Syracuse University and received
her Ph.D. from Princeton University.
Katharine E. Spink, Ph.D., has served as our Vice President of Operations, Regenerative Medicine
Programs since February 2009. From January 2008 until February 2009, Dr. Spink
served as our Senior Director of Regenerative Medicine Program Operations. From
January 2007 until January 2008, Dr. Spink served as our Program Director for
Cardiovascular Disease. Dr. Spink joined Geron in December 2003, and served
various roles within our Corporate Development group until January 2007. Prior
to Geron, Dr. Spink was with the global management consulting firm McKinsey
& Company, where she advised clients in the biotechnology, pharmaceutical,
and medical device industries on matters relating to R&D strategy, business
development, and marketing. Dr. Spink holds a B.A. in biochemistry from Rice
University and a Ph.D. in cancer biology from Stanford University.
Employees
As of December 31, 2009, we had 172 employees of whom 50 hold Ph.D.
degrees and 44 hold other advanced degrees. Of our total workforce, 144
employees were engaged in, or directly support, our research and development
activities and 28 employees were engaged in business development, legal, finance
and administration. We also retain outside consultants. None of our employees
are covered by a collective bargaining agreement, nor have we experienced work
stoppages. We consider relations with our employees to be good.
ITEM 1A. RISK FACTORS
Our business is subject to various risks, including those described
below. You should carefully consider these risk factors, together with all of
the other information included in this Form 10-K. Any of these risks could
materially adversely affect our business, operating results and financial
condition.
19
RISKS RELATED TO OUR
BUSINESS
Our business is at an early stage of
development.
Our business is at an early stage of development, in that we do not yet
have product candidates in late-stage clinical trials or on the market. We have
sponsored six Phase I or I/II trials of our lead anti-cancer drug, imetelstat,
in patients with chronic lymphoproliferative diseases, solid tumor malignancies,
non-small cell lung cancer, breast cancer and multiple myeloma. Four of those
trials have completed patient enrollment and the remaining two are expected to
complete enrollment in 2010. We currently plan to advance imetelstat to Phase II
trials in 2010 in four different malignancies. Patient enrollment for our
telomerase cancer vaccine, GRNVAC1, in patients with acute myelogenous leukemia
is now complete and we are awaiting the results from this Phase II trial. We
have no other product candidates in clinical testing. Currently, the clinical
trial of GRNOPC1, our human embryonic stem cell (hESC)-derived therapy targeted
for the treatment of acute spinal cord injury, has been placed on clinical hold
by the U.S. Food and Drug Administration (FDA). We are in discussions with the
FDA to answer its questions and seek the release of the clinical hold to permit
us to proceed with the clinical trial. Our ability to develop product candidates
that progress to and through clinical trials is subject to our ability to, among
other things:
- succeed in our research and
development efforts;
- select therapeutic compounds or
cell therapies for development;
- obtain required regulatory
approvals, including release by the FDA of the clinical hold for GRNOPC1;
- manufacture product candidates;
and
- collaborate successfully with clinical trial sites, academic institutions, physician investigators, clinical research organizations and other third parties.
Potential lead drug compounds or other product candidates and
technologies require significant preclinical and clinical testing prior to
regulatory approval in the United States and other countries. Our product
candidates may prove to have undesirable and unintended side effects or other
characteristics adversely affecting their safety, efficacy or cost-effectiveness
that could prevent or limit their commercial use. In addition, our product
candidates may not prove to be more effective for treating disease or injury
than current therapies. Accordingly, we may have to delay or abandon efforts to
research, develop or obtain regulatory approvals to market our product
candidates. In addition, we will need to determine whether any of our potential
products can be manufactured in commercial quantities at an acceptable cost. Our
research and development efforts may not result in a product that can be or will
be approved by regulators or marketed successfully. Competitors may have
proprietary rights which prevent us from developing and marketing our products
or they may sell similar, superior or lower-cost products. Because of the
significant scientific, regulatory and commercial milestones that must be
reached for any of our development programs or product candidates to be
successful, any program or product candidate may be abandoned, even after we
have expended significant resources, such as our investments in telomerase
technology, hESCs, imetelstat, GRNVAC1 and GRNOPC1, which could adversely affect
our business and cause a sharp drop in our stock price.
The science and technology of telomere biology and telomerase and hESCs
are relatively new. There is no precedent for the successful commercialization
of therapeutic product candidates based on our technologies. These development
programs are therefore particularly risky. In addition, we, our licensees or our
collaborators must undertake significant research and development activities to
develop product candidates based on our technologies, which will require
additional funding and may take years to accomplish, if ever.
Restrictions on the use of hESCs, political commentary and the ethical
and social implications of research involving hESCs could prevent us from
developing or gaining acceptance for commercially viable products based upon
such stem cells and adversely affect the market price of our common
stock.
Some of our most important programs involve the use of stem cells that
are derived from human embryos. The use of hESCs gives rise to ethical and
social issues regarding the appropriate use of these cells. Our research related
to hESCs may become the subject of adverse commentary or publicity, which could
significantly harm the market price for our common stock.
20
Some political and religious groups have voiced opposition to our
technology and practices. We use stem cells derived from human embryos that had
been created for in vitro fertilization procedures but were no longer
desired or suitable for that use and were donated with appropriate informed
consent. Many research institutions, including some of our scientific
collaborators, have adopted policies regarding the ethical use of human
embryonic tissue. These policies may have the effect of limiting the scope of
research conducted using hESCs, thereby impairing our ability to conduct
research in this field.
Furthermore, on March 9, 2009, President Obama issued Executive Order
13505, entitled “Removing Barriers to Responsible Scientific Research Involving
Human Stem Cells” (the Executive Order). As a result, in July 2009 the Secretary
of Health and Human Services, through the Director of the National Institutes of
Health (NIH), issued new guidelines relating to human stem cell research. Under
the new guidelines, federal funding is allowed for research using hESCs derived
from embryos created by in vitro
fertilization for reproductive purposes, but are no longer needed for that
purpose. Strict ethics requirements must be followed to qualify new stem cell
lines, including extensive documentation around consent forms and written
policies and procedures. Certain states are considering enacting, or already
have enacted, legislation relating to stem cell research, including California,
whose voters approved Proposition 71 to provide state funds for stem cell
research in November 2004. In the United Kingdom and other countries, the use of
embryonic or fetal tissue in research (including the derivation of hESCs) is
regulated by the government, whether or not the research involves government
funding.
Government-imposed restrictions with respect to use of embryos or hESCs
in research and development could have a material adverse effect on us,
including:
- harming our ability to establish
critical partnerships and collaborations;
- delaying or preventing progress in
our research, product development or clinical testing;
- preventing commercialization of
therapies derived from hESCs; and
- as a result of the potential adverse effects above, causing a decrease in the price of our stock.
RISKS RELATED TO OUR FINANCIAL POSITION
AND
NEED FOR ADDITIONAL FINANCING
NEED FOR ADDITIONAL FINANCING
We have a history of losses and anticipate future losses, and continued
losses could impair our ability to sustain
operations.
We have incurred operating losses every year since our operations began
in 1990. As of December 31, 2009, our accumulated deficit was approximately
$577.3 million. Losses have resulted principally from costs incurred in
connection with our research and development activities and from general and
administrative costs associated with our operations. We expect to incur
additional operating losses and, as our development efforts and clinical testing
activities continue, our operating losses may increase in size.
Substantially all of our revenues to date have been research support
payments under collaboration agreements and revenues from our licensing
arrangements. We may be unsuccessful in entering into any new corporate
collaboration or license agreement that results in revenues. We do not expect
that the revenues generated from these arrangements will be sufficient alone to
continue or expand our research or development activities and otherwise sustain
our operations.
While we receive royalty revenue from licenses, we do not currently
expect to receive sufficient royalty revenues from these licenses to
independently sustain our operations. Our ability to continue or expand our
research and development activities and otherwise sustain our operations is
dependent on our ability, alone or with others, to, among other things,
manufacture and market therapeutic products.
We also expect to experience negative cash flow for the foreseeable
future as we fund our operating losses and capital expenditures. This will
result in decreases in our working capital, total assets and stockholders’
equity, which may not be offset by future financings. We will need to generate
significant revenues to achieve profitability. We may not be able to generate
these revenues, and we may never achieve profitability. Our failure to achieve
profitability could negatively impact the market price of our common stock. Even
if we do become profitable, we cannot assure you that we would be able to
sustain or increase profitability on a quarterly or annual basis.
21
We will need additional capital to conduct our operations and develop our
product candidates, and our ability to obtain the necessary funding is
uncertain.
We will require substantial capital resources in order to conduct our
operations and develop our product candidates, and we cannot assure you that our
existing capital resources, interest income and equipment financing arrangement
will be sufficient to fund future planned operations. The timing and degree of
any future capital requirements will depend on many factors,
including:
- the accuracy of the assumptions
underlying our estimates for our capital needs for the 2010 fiscal year
and beyond;
- the magnitude and scope of our
research and development programs;
- the progress we make in our
research and development programs, preclinical development and clinical
trials;
- our ability to establish, enforce
and maintain strategic arrangements for research, development, clinical
testing, manufacturing and
marketing;
- the number and type of product
candidates that we pursue;
- the time and costs involved in
obtaining regulatory approvals and clearances; and
- the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims.
We do not have any committed sources of capital. Additional financing
through strategic collaborations, public or private equity financings, capital
lease transactions or other financing sources may not be available on acceptable
terms, or at all. The receptivity of the public and private equity markets to
proposed financings is substantially affected by the general economic, market
and political climate and by other factors which are unpredictable and over
which we have no control. Additional equity financings, if we obtain them, could
result in significant dilution to stockholders. Further, in the event that
additional funds are obtained through arrangements with collaborative partners,
these arrangements may require us to relinquish rights to some of our
technologies, product candidates or proposed products that we would otherwise
seek to develop and commercialize ourselves. If sufficient capital is not
available, we may be required to delay, reduce the scope of or eliminate one or
more of our programs, any of which could have a material adverse effect on our
business.
RISKS RELATED TO CLINICAL AND
COMMERCIALIZATION ACTIVITIES
Delays in the commencement of clinical testing of our current and
potential product candidates could result in increased costs to us and delay our ability to generate revenues.
The commencement of clinical trials
can be delayed for a variety of reasons, including delays in:
- demonstrating sufficient safety
and efficacy to obtain regulatory clearance to commence a clinical
trial;
- manufacturing sufficient
quantities or producing drugs meeting our quality standards of a product
candidate;
- obtaining approval of an
Investigational New Drug (IND) application or proposed trial design from the
FDA;
- reaching agreement on acceptable
terms with our collaborators on all aspects of the clinical trial,
including the contract research
organizations (CROs) and the trial sites; and
- obtaining institutional review board approval to conduct a clinical trial at a prospective site.
In addition, clinical trials may be delayed due to insufficient patient
enrollment, which is a function of many factors, including the size and nature
of the patient population, the nature of the protocol, the proximity of patients
to clinical sites, the availability of effective treatments for the relevant
disease, and the eligibility criteria for the clinical trial. Delays in
commencing clinical testing of our product candidates could have a material
adverse effect on our business.
22
We do not have experience as a company conducting large-scale clinical
trials, or in other areas required for the successful commercialization and
marketing of our product candidates.
We have no experience as a company in conducting large-scale, late stage
clinical trials. We cannot be certain that planned clinical trials will begin or
be completed on time, if at all. Large-scale trials would require either
additional financial and management resources, or reliance on third-party
clinical investigators, CROs or consultants. Relying on third-party clinical
investigators or CROs may force us to encounter delays that are outside of our
control. Any such delays could have a material adverse effect on our
business.
We also do not currently have marketing and distribution capabilities for
our product candidates. Developing an internal sales and distribution capability
would be an expensive and time-consuming process. We may enter into agreements
with third parties that would be responsible for marketing and distribution.
However, these third parties may not be capable of successfully selling any of
our product candidates. The inability to commercialize and market our product
candidates could materially adversely affect our business.
Obtaining regulatory approvals to market our product candidates in the
United States and other countries is a costly and lengthy process and we cannot
predict whether or when we will be permitted to commercialize our product
candidates.
Federal, state and local governments in the United States and governments
in other countries have significant regulations in place that govern many of our
activities and may prevent us from creating commercially viable products from
our discoveries. The regulatory process, particularly for biopharmaceutical
product candidates like ours, is uncertain, can take many years and requires the
expenditure of substantial resources.
Our potential product candidates will require extensive preclinical and
clinical testing prior to submission of any regulatory application to commence
commercial sales. In particular, human pharmaceutical therapeutic product
candidates are subject to rigorous requirements of the FDA in the United States
and similar health authorities in other countries in order to demonstrate safety
and efficacy. Data obtained from preclinical and clinical activities is
susceptible to varying interpretations that could delay, limit or prevent
regulatory agency approvals. In addition, delays or rejections may be
encountered as a result of changes in regulatory agency policy during the period
of product development and/or the period of review of any application for
regulatory agency approval for a product candidate.
Any product candidate that we or our collaborators develop must receive
all relevant regulatory agency approvals before it may be marketed in the United
States or other countries. Obtaining regulatory approval is a lengthy, expensive
and uncertain process. Because certain of our product candidates involve the
application of new technologies or are based upon a new therapeutic approach,
they may be subject to substantial additional review by various government
regulatory authorities, and, as a result, the process of obtaining regulatory
approvals for them may proceed more slowly than for product candidates based
upon more conventional technologies.
Delays in obtaining regulatory
agency approvals could:
- significantly harm the marketing
of any products that we or our collaborators develop;
- impose costly procedures upon our
activities or the activities of our collaborators;
- diminish any competitive
advantages that we or our collaborators may attain; or
- adversely affect our ability to receive royalties and generate revenues and profits.
Even if we commit the
necessary time and resources, the required regulatory agency approvals may not
be obtained for any product candidates developed by us or in collaboration with
us. If we obtain regulatory agency approval for a new product, this approval may
entail limitations on the indicated uses for which it can be marketed that could
limit the potential commercial use of the product.
23
Failure to achieve continued compliance with government regulation over
approved products could delay or halt commercialization of our products.
Approved products and their manufacturers are subject to continual
review, and discovery of previously unknown problems with a product or its
manufacturer may result in restrictions on the product or manufacturer,
including withdrawal of the product from the market. The sale by us or our
collaborators of any commercially viable product will be subject to government
regulation from several standpoints, including the processes of:
- manufacturing;
- advertising and
promoting;
- selling and
marketing;
- labeling; and
- distribution.
If, and to the extent
that, we are unable to comply with these regulations, our ability to earn
revenues will be materially and negatively impacted.
Failure to comply with regulatory requirements can result in severe civil
and criminal penalties, including but not limited to:
- recall or seizure of
products;
- injunction against the
manufacture, distribution and sales and marketing of products;
and
- criminal prosecution.
The imposition of any of
these penalties or other commercial limitations could significantly impair our
business, financial condition and results of operations.
RISKS RELATED TO PROTECTING OUR INTELLECTUAL
PROPERTY
Impairment of our intellectual property rights may adversely affect the
value of our technologies and product candidates and limit our ability to pursue
their development.
Protection of our proprietary technology is critically important to our
business. Our success will depend in part on our ability to obtain and enforce
our patents and maintain trade secrets, both in the United States and in other
countries. Further, our patents may be challenged, invalidated or circumvented,
and our patent rights may not provide proprietary protection or competitive
advantages to us. In the event that we are unsuccessful in obtaining and
enforcing patents, our business would be negatively impacted.
The patent positions of pharmaceutical and biopharmaceutical companies,
including ours, are highly uncertain and involve complex legal and technical
questions. In particular, legal principles for biotechnology patents in the
United States and in other countries are evolving, and the extent to which we
will be able to obtain patent coverage to protect our technology, or enforce
issued patents, is uncertain. In the United States, recent court decisions in
patent cases as well as proposed legislative changes to the patent system only
exacerbate this uncertainty. Furthermore, significant amendments to the
regulations governing the process of obtaining patents were proposed in a new
rule package by the United States Patent and Trademark Office (the Patent
Office) in 2007. The proposed new rules were widely regarded as detrimental to
the interests of biotechnology and pharmaceutical companies. The implementation
of the rule package was blocked by a court injunction requested by a
pharmaceutical company. The Patent Office challenged the court decision through
an appeal to the U.S. Court of Appeals for the Federal Circuit (CAFC), but the
appeal was dismissed in November 2009, after the Patent Office changed course
and rescinded the proposed new rules. At this point we do not know whether the
Patent Office will attempt to introduce new rules to replace those that were
recently withdrawn or whether any such new rules would also be challenged.
In Europe, the European Patent Convention prohibits the granting of
European patents for inventions that concern “uses of human embryos for
industrial or commercial purposes.” The European Patent Office (EPO) was earlier
interpreting this prohibition broadly, and applying it to reject claims in any
patent application that pertained to hESCs. An early patent application filed by
the Wisconsin Alumni Research
24
Foundation (WARF) with
claims covering the original isolation of hESCs was appealed as a test case, and
examination of other hESC patent applications was suspended while that case was
heard. In November 2008, the EPO Enlarged Board of Appeals held that the claims
in the WARF application were unpatentable. Geron holds a worldwide license under
this patent family, and since the decision is not subject to further appeal,
this WARF patent family will not afford protection to Geron’s hESC-based product
candidates in Europe. However, the reason given by the EPO for the decision was
narrowly focused: the EPO found the claims objectionable on the basis that at
the time that WARF filed the patent application it was necessary to use a human
embryo to obtain hESCs since no cell lines were available. In contrast, the
hESCs that we use, and which we employed in the technologies claimed in our own
European patent applications, were sourced from established hESC lines.
Consequently, the decision in the WARF case does not directly address the
patentability of the subject matter in our filings. The EPO has recently
restarted examination of hESC patent applications, but is being inconsistent in
its application of the WARF decision to these later filed cases. At this time,
we do not know whether or to what extent we will be able to obtain patent
protection for our hESC technologies in Europe. If we are unable to protect our
inventions related to hESCs in Europe, our business would be negatively
impacted.
Challenges to our patent rights can result in costly and time-consuming
legal proceedings that may prevent or limit development of our product
candidates.
Publication of discoveries in scientific or patent literature tends to
lag behind actual discoveries by at least several months and sometimes several
years. Therefore, the persons or entities that we or our licensors name as
inventors in our patents and patent applications may not have been the first to
invent the inventions disclosed in the patent applications or patents, or the
first to file patent applications for these inventions. As a result, we may not
be able to obtain patents for discoveries that we otherwise would consider
patentable and that we consider to be extremely significant to our future
success.
Where several parties seek U.S. patent protection for the same
technology, the Patent Office may declare an interference proceeding in order to
ascertain the party to which the patent should be issued. Patent interferences
are typically complex, highly contested legal proceedings, subject to appeal.
They are usually expensive and prolonged, and can cause significant delay in the
issuance of patents. Moreover, parties that receive an adverse decision in an
interference can lose important patent rights. Our pending patent applications,
or our issued patents, may be drawn into interference proceedings which may
delay or prevent the issuance of patents, or result in the loss of issued patent
rights. By way of example, we are currently a party to an interference
proceeding that involves patent filings for making endoderm cells from hESCs. We
requested that the Patent Office declare this interference after Novocell Inc.
was granted patent claims that conflict with subject matter we filed in an
earlier patent application. The interference proceeding will determine whether
we or Novocell are entitled to such patent claims. Since this interference is
still ongoing, we cannot predict what the outcome will be.
Outside of the United States, certain jurisdictions, such as Europe, New
Zealand and Australia, permit oppositions to be filed against the granting of
patents. Because our intent is to commercialize products internationally,
securing both proprietary protection and freedom to operate outside of the
United States is important to our business. We are involved in both opposing the
grant of patents to others through such opposition proceedings and in defending
our patent applications against oppositions filed by others. For example, we
have been involved in two patent oppositions before the EPO with a Danish
company, Pharmexa. Pharmexa (which acquired the Norwegian company GemVax in
2005) was developing a cancer vaccine that employs a short telomerase peptide to
induce an immune response against telomerase and was conducting a Phase III
clinical trial. Pharmexa obtained a European patent with broad claims to the use
of telomerase vaccines for the treatment of cancer, and Geron opposed that
patent in 2004. In 2005, the Opposition Division (OD) of the EPO revoked the
claims originally granted to Pharmexa, but permitted Pharmexa to add new,
narrower claims limited to five specific small peptide fragments of telomerase.
The decision was appealed to the Technical Board of Appeals (TBA). In August
2007, the TBA ruled, consistent with the decision of the OD, that Pharmexa was
not entitled to the originally granted broad claims but was only entitled to the
narrow claims limited to the five small peptides.
In parallel, Pharmexa opposed a European patent held by Geron, the claims
of which cover many facets of human telomerase, including the use of telomerase
peptides in cancer vaccines. In June 2006, the OD of the EPO revoked three of
the granted claims in Geron’s patent, specifically the three claims covering
telomerase
25
peptide cancer vaccines.
We have appealed that decision to the TBA, and that appeal is still pending.
Because this appeal is ongoing, the outcome cannot be determined at this time.
We are also seeking to obtain patent coverage in Europe for telomerase peptides
through a European divisional patent application. If and when those patent
claims are issued, they too may be subject to an opposition proceeding. In late
2008, Pharmexa reported that it sold its telomerase vaccine program to a Korean
company, KAEL Co. Ltd.
European opposition and appeal proceedings can take several years to
reach final decision. The oppositions discussed above reflect the complexity of
the patent landscape in which we operate, and illustrate the risks and
uncertainties. We are also currently involved in other patent opposition
proceedings in Europe and Australia.
Patent opposition proceedings are not currently available in the U.S.
patent system, but legislation has been proposed to introduce them. However,
issued U.S. patents can be reexamined by the Patent Office at the request of a
third party. Patents owned or licensed by Geron may therefore be subject to
reexamination. As in any legal proceeding, the outcome of patent reexaminations
is uncertain, and a decision adverse to our interests could result in the loss
of valuable patent rights.
In July 2006, requests were filed on behalf of the Foundation for
Taxpayer and Consumer Rights (now renamed as “Consumer Watchdog”) for
reexamination of three issued U.S. patents owned by WARF and relating to hESCs.
These three patents (U.S. Patent Nos. 5,843,780, 6,200,806 and 7,029,913), which
are the U.S. equivalents of the European WARF case discussed above, are licensed
to Geron pursuant to a January 2002 license agreement with WARF. The license
agreement conveys exclusive rights to Geron under the WARF patents for the
development and commercialization of therapeutics based on neural cells,
cardiomyocytes and pancreatic islet cells, derived from hESCs, as well as
non-exclusive rights for other product opportunities. In October 2006, the
Patent Office initiated the reexamination proceedings. After initially rejecting
the patent claims, the Patent Office recently issued decisions in all three
cases upholding the patentability of the claims. The decisions to uphold the
5,843,780 and 6,200,806 patents are final and not subject to further appeal.
Consumer Watchdog appealed the decision on the 7,029,913 patent. We cooperated
with WARF in these reexamination actions and expect that WARF will continue to
vigorously defend its patent position in this appeal. While the decisions in
these reexamination proceedings to date have all been favorable to our patent
position, the outcome of the appeal or of any future reexamination proceedings
cannot be determined at this time. Reduction or loss of claim scope in these
WARF embryonic stem cell patents would negatively impact Geron’s proprietary
position in this technology.
As more groups become engaged in scientific research and product
development in the areas of telomerase biology and embryonic stem cells, the
risk of our patents being challenged through patent interferences, oppositions,
reexaminations or other means will likely increase. Challenges to our patents
through these procedures can be extremely expensive and time-consuming, even if
the outcome is favorable to us. An adverse outcome in a patent dispute could
severely harm our business by:
- causing us to lose patent rights
in the relevant jurisdiction(s);
- subjecting us to litigation, or
otherwise preventing us from commercializing potential products in the relevant
jurisdiction(s);
- requiring us to obtain licenses to
the disputed patents;
- forcing us to cease using the
disputed technology; or
- requiring us to develop or obtain alternative technologies.
Furthermore, if such
challenges to our patent rights are not resolved promptly in our favor, our
existing business relationships may be jeopardized and we could be delayed or
prevented from entering into new collaborations or from commercializing certain
products, which could materially harm our business.
If we fail to meet our obligations under license agreements, we may lose
our rights to key technologies on which our business depends.
Our business depends on several critical technologies that are based in
part on patents licensed from third parties. Those third-party license
agreements impose obligations on us, such as payment obligations and obligations
to diligently pursue development of commercial products under the licensed
patents. If a
26
licensor believes that
we have failed to meet our obligations under a license agreement, the licensor
could seek to limit or terminate our license rights, which could lead to costly
and time-consuming litigation and, potentially, a loss of the licensed rights.
During the period of any such litigation our ability to carry out the
development and commercialization of potential products could be significantly
and negatively affected. If our license rights were restricted or ultimately
lost, our ability to continue our business based on the affected technology
would be severely adversely affected.
We may be subject to litigation that will be costly to defend or pursue
and uncertain in its outcome.
Our business may bring us into conflict with our licensees, licensors, or
others with whom we have contractual or other business relationships, or with
our competitors or others whose interests differ from ours. If we are unable to
resolve those conflicts on terms that are satisfactory to all parties, we may
become involved in litigation brought by or against us. That litigation is
likely to be expensive and may require a significant amount of management’s time
and attention, at the expense of other aspects of our business. The outcome of
litigation is always uncertain, and in some cases could include judgments
against us that require us to pay damages, enjoin us from certain activities, or
otherwise affect our legal or contractual rights, which could have a significant
adverse effect on our business.
We may be subject to infringement claims that are costly to defend, and
which may limit our ability to use disputed technologies and prevent us from
pursuing research and development or commercialization of potential
products.
Our commercial success depends significantly on our ability to operate
without infringing patents and the proprietary rights of others. Our
technologies may infringe the patents or proprietary rights of others. In
addition, we may become aware of discoveries and technology controlled by third
parties that are advantageous to our programs. In the event our technologies
infringe the rights of others or we require the use of discoveries and
technology controlled by third parties, we may be prevented from pursuing
research, development or commercialization of potential products or may be
required to obtain licenses to those patents or other proprietary rights or
develop or obtain alternative technologies. We have obtained licenses from
several universities and companies for technologies that we anticipate
incorporating into our potential products, and we initiate negotiation for
licenses to other technologies as the need or opportunity arises. We may not be
able to obtain a license to patented technology on commercially favorable terms,
or at all. If we do not obtain a necessary license, we may need to redesign our
technologies or obtain rights to alternate technologies, the research and
adoption of which could cause delays in product development. In cases where we
are unable to license necessary technologies, we could be prevented from
developing certain potential products. Our failure to obtain alternative
technologies or a license to any technology that we may require to research,
develop or commercialize our product candidates would significantly and
negatively affect our business.
Much of the information and know-how that is critical to our business is
not patentable and we may not be able to prevent others from obtaining this
information and establishing competitive enterprises.
We sometimes rely on trade secrets to protect our proprietary technology,
especially in circumstances in which we believe patent protection is not
appropriate or available. We attempt to protect our proprietary technology in
part by confidentiality agreements with our employees, consultants,
collaborators and contractors. We cannot assure you that these agreements will
not be breached, that we would have adequate remedies for any breach, or that
our trade secrets will not otherwise become known or be independently discovered
by competitors, any of which would harm our business significantly.
RISKS RELATED TO OUR RELATIONSHIPS WITH THIRD
PARTIES
We depend on other parties to help us develop, manufacture and test our
product candidates, and our ability to develop and commercialize potential
products may be impaired or delayed if collaborations are
unsuccessful.
Our strategy for the development, clinical testing and commercialization
of our product candidates requires that we enter into collaborations with
corporate partners, licensors, licensees and others. We are dependent upon the
subsequent success of these other parties in performing their respective
27
responsibilities and the
continued cooperation of our partners. By way of examples: Merck is developing
cancer vaccines targeted to telomerase other than dendritic cell-based vaccines;
Sienna is developing cancer diagnostics using our telomerase technology; and GE
Healthcare is developing cell-based assays using cells derived from our hESCs.
Our collaborators may not cooperate with us or perform their obligations under
our agreements with them. We cannot control the amount and timing of our
collaborators’ resources that will be devoted to activities related to our
collaborative agreements with them. Our collaborators may choose to pursue
existing or alternative technologies in preference to those being developed in
collaboration with us.
Under agreements with other parties,
we may rely significantly on them to, among other activities:
- conduct research and development
activities in conjunction with us;
- design and conduct advanced
clinical trials in the event that we reach clinical trials;
- fund research and development
activities with us;
- manage and license certain patent
rights;
- pay us fees upon the achievement
of milestones; and
- market with us any commercial products that result from our collaborations.
The development and commercialization of potential products will be
delayed if collaborators or other partners fail to conduct these activities in a
timely manner or at all. In addition, our collaborators could terminate their
agreements with us and we may not receive any development or milestone payments.
If we do not achieve milestones set forth in the agreements, or if our
collaborators breach or terminate their collaborative agreements with us, our
business may be materially harmed.
We also rely on other companies for certain process development,
manufacturing or other technical scientific work, especially with respect to our
imetelstat, GRNVAC1, GRNOPC1 and GRNCM1 programs. We have contracts with these
companies that specify the work to be done and results to be achieved, but we do
not have direct control over their personnel or operations. If these companies
do not perform the work which they were assigned, our ability to develop or
manufacture our product candidates could be significantly harmed.
Our reliance on the activities of our non-employee consultants, research
institutions, and scientific contractors, whose activities are not wholly within
our control, may lead to delays in development of our product
candidates.
We rely extensively upon and have relationships with scientific
consultants at academic and other institutions, some of whom conduct research at
our request, and other consultants who assist us in formulating our research and
development and clinical strategy or other matters. These consultants are not
our employees and may have commitments to, or consulting or advisory contracts
with, other entities that may limit their availability to us. We have limited
control over the activities of these consultants and, except as otherwise
required by our collaboration and consulting agreements, can expect only limited
amounts of their time to be dedicated to our activities.
In addition, we have formed research collaborations with many academic
and other research institutions throughout the world. These research facilities
may have commitments to other commercial and noncommercial entities. We have
limited control over the operations of these laboratories and can expect only
limited amounts of their time to be dedicated to our research
goals.
If any of these third parties are unable or refuse to contribute to
projects on which we need their help, our ability to generate advances in our
technologies and develop our product candidates could be significantly
harmed.
28
RISKS RELATED TO COMPETITIVE
FACTORS
The loss of key personnel could slow our ability to conduct research and
develop product candidates.
Our future success depends to a significant extent on the skills,
experience and efforts of our executive officers and key members of our
scientific staff. We face intense competition for qualified individuals from
numerous pharmaceutical, biopharmaceutical and biotechnology companies, as well
as academic and other research institutions. We may be unable to retain our
current personnel or attract or assimilate other highly qualified management and
scientific personnel in the future on acceptable terms. The loss of any or all
of these individuals could harm our business and might significantly delay or
prevent the achievement of research, development or business
objectives.
Our product candidates are likely to be expensive to manufacture, and
they may not be profitable if we are unable to significantly reduce the costs to
manufacture them.
Our telomerase inhibitor compound, imetelstat, our telomerase cancer
vaccine, GRNVAC1, and our hESC-based products are likely to be more expensive to
manufacture than most other drugs currently on the market today.
Oligonucleotides are relatively large molecules with complex chemistry, and the
cost of manufacturing an oligonucleotide like imetelstat is greater than the
cost of making most small-molecule drugs. Our present manufacturing processes
are conducted at a modest scale and we hope to substantially reduce
manufacturing costs through process improvements, as well as through scale
increases. If we are not able to do so, however, and, depending on the pricing
of the potential product, the profit margin on the telomerase inhibitor may be
significantly less than that of most drugs on the market today.
GRNVAC1 is an autologous therapy that is produced from a patient’s blood
using a unique process that generates highly activated dendritic cells that
contain RNA coding for the protein component of telomerase. If we are unable to
scalably produce dendritic cells at a lower manufacturing cost, the cost of
GRNVAC1 may reduce the affordability of the therapy for patients and reduce our
potential profitability.
Our manufacturing processes for differentiated cells from hESCs are
conducted at a small scale and at a high cost per unit measure. The cell-based
therapies we are developing based on hESCs will probably require large
quantities of cells. We continue to develop processes to scale up production of
the cells in a cost-effective way. We may not be able to charge a high enough
price for any cell therapy product we develop, even if it is safe and effective,
to make a profit. If we are unable to realize significant profits from our
potential product candidates, our business would be materially
harmed.
Some of our competitors may develop technologies that are superior to or
more cost-effective than ours, which may impact the commercial viability of our
technologies and which may significantly damage our ability to sustain
operations.
The pharmaceutical and biotechnology industries are intensely
competitive. Other pharmaceutical and biotechnology companies and research
organizations currently engage in or have in the past engaged in efforts related
to the biological mechanisms that are the focus of our programs in oncology and
human embryonic stem cell therapies, including the study of telomeres,
telomerase and hESCs. In addition, other products and therapies that could
compete directly with the product candidates that we are seeking to develop and
market currently exist or are being developed by pharmaceutical and
biopharmaceutical companies and by academic and other research
organizations.
Many companies are developing alternative therapies to treat cancer and,
in this regard, are competitors of ours. According to public data from the FDA
and NIH, there are more than 200 approved anti-cancer products on the market in
the United States, and several thousand in clinical development.
Many of the pharmaceutical companies developing and marketing these
competing products (including GlaxoSmithKline, Bristol-Myers Squibb Company and
Novartis AG, among others) have significantly greater financial resources and
expertise than we do in:
- research and
development;
- manufacturing;
- preclinical and clinical testing;
29
- obtaining regulatory approvals;
and
- marketing and distribution.
Smaller companies may also prove to be significant competitors,
particularly through collaborative arrangements with large and established
companies. Academic institutions, government agencies and other public and
private research organizations may also conduct research, seek patent protection
and establish collaborative arrangements for research, clinical development and
marketing of products similar to ours. These companies and institutions compete
with us in recruiting and retaining qualified scientific and management
personnel as well as in acquiring technologies complementary to our
programs.
In addition to the above factors, we
expect to face competition in the following areas:
- product efficacy and
safety;
- the timing and scope of regulatory
consents;
- availability of
resources;
- reimbursement
coverage;
- price; and
- patent position, including potentially dominant patent positions of others.
As a result of the
foregoing, our competitors may develop more effective or more affordable
products, or achieve earlier patent protection or product commercialization than
we do. Most significantly, competitive products may render any product
candidates that we develop obsolete, which would negatively impact our business
and ability to sustain operations.
To be successful, our product candidates must be accepted by the health
care community, which can be very slow to adopt or unreceptive to new
technologies and products.
Our product candidates and those developed by our collaborators, if
approved for marketing, may not achieve market acceptance since hospitals,
physicians, patients or the medical community in general may decide not to
accept and utilize these products. The product candidates that we are attempting
to develop represent substantial departures from established treatment methods
and will compete with a number of conventional drugs and therapies manufactured
and marketed by major pharmaceutical companies. The degree of market acceptance
of any of our developed potential products will depend on a number of factors,
including:
- our establishment and
demonstration to the medical community of the clinical efficacy and
safety of our product
candidates;
- our ability to create products
that are superior to alternatives currently on the market;
- our ability to establish in the
medical community the potential advantage of our treatments over alternative treatment methods;
and
- reimbursement policies of government and third-party payors.
If the health care
community does not accept our potential products for any of the foregoing
reasons, or for any other reason, our business would be materially
harmed.
If we fail to obtain acceptable prices or adequate reimbursement for our
product candidates, the use of our potential products could be severely
limited.
Our ability to successfully commercialize our product candidates will
depend significantly on our ability to obtain acceptable prices and the
availability of reimbursement to the patient from third-party payors.
Significant uncertainty exists as to the reimbursement status of newly-approved
health care products, including pharmaceuticals. If our potential products are
not considered cost-effective or if we fail to generate adequate third-party
reimbursement for the users of our potential products and treatments, then we
may be unable to maintain price levels sufficient to realize an appropriate
return on
30
our investment for
potential products currently in development. In both U.S. and other markets,
sales of our potential products, if any, will depend in part on the availability
of reimbursement from third-party payors, examples of which
include:
- government health administration
authorities;
- private health
insurers;
- health maintenance organizations;
and
- pharmacy benefit management companies.
Both federal and state governments in the United States and governments
in other countries continue to propose and pass legislation designed to contain
or reduce the cost of health care. Legislation and regulations affecting the
pricing of pharmaceuticals and other medical products may be adopted before any
of our potential products are approved for marketing. Cost control initiatives
could decrease the price that we receive for any product candidate we may
develop in the future. In addition, third-party payors are increasingly
challenging the price and cost-effectiveness of medical products and services
and any of our potential products may ultimately not be considered
cost-effective by these third parties. Any of these initiatives or developments
could materially harm our business.
RISKS RELATED TO ENVIRONMENTAL AND PRODUCT
LIABILITY
Our activities involve hazardous materials, and improper handling of
these materials by our employees or agents could expose us to significant legal
and financial penalties.
Our research and development activities involve the controlled use of
hazardous materials, chemicals and various radioactive compounds. As a
consequence, we are subject to numerous environmental and safety laws and
regulations, including those governing laboratory procedures, exposure to
blood-borne pathogens and the handling of biohazardous materials. We may be
required to incur significant costs to comply with current or future
environmental laws and regulations and may be adversely affected by the cost of
compliance with these laws and regulations.
Although we believe that our safety procedures for using, handling,
storing and disposing of hazardous materials comply with the standards
prescribed by state and federal regulations, the risk of accidental
contamination or injury from these materials cannot be eliminated. In the event
of such an accident, state or federal authorities could curtail our use of these
materials and we could be liable for any civil damages that result, the cost of
which could be substantial. Further, any failure by us to control the use,
disposal, removal or storage, or to adequately restrict the discharge, or assist
in the clean up, of hazardous chemicals or hazardous, infectious or toxic
substances could subject us to significant liabilities, including joint and
several liability under certain statutes. Any such liability could exceed our
resources and could have a material adverse effect on our business, financial
condition and results of operations. Additionally, an accident could damage our
research and manufacturing facilities and operations.
Additional federal, state and local laws and regulations affecting us may
be adopted in the future. We may incur substantial costs to comply with these
laws and regulations and substantial fines or penalties if we violate any of
these laws or regulations, which would adversely affect our
business.
We may not be able to obtain or maintain sufficient insurance on
commercially reasonable terms or with adequate coverage against potential
liabilities in order to protect ourselves against product liability
claims.
Our business exposes us to potential product liability risks that are
inherent in the testing, manufacturing and marketing of human therapeutic and
diagnostic products. We may become subject to product liability claims if the
use of our potential products is alleged to have injured subjects or patients.
This risk exists for product candidates tested in human clinical trials as well
as potential products that are sold commercially. We currently have limited
clinical trial liability insurance and we may not be able to maintain this type
of insurance for any of our clinical trials. In addition, product liability
insurance is becoming increasingly expensive. Being unable to obtain or maintain
product liability insurance in the future on acceptable terms or with adequate
coverage against potential liabilities could have a material adverse effect on
our business.
31
RISKS RELATED TO OUR COMMON STOCK AND
FINANCIAL REPORTING
Our stock price has historically been very
volatile.
Stock prices and trading volumes for many biopharmaceutical companies
fluctuate widely for a number of reasons, including factors which may be
unrelated to their businesses or results of operations such as media coverage,
legislative and regulatory measures and the activities of various interest
groups or organizations. This market volatility, as well as general domestic or
international economic, market and political conditions, could materially and
adversely affect the market price of our common stock and the return on your
investment.
Historically, our stock price has been extremely volatile. Between
January 2000 and December 2009, our stock has traded as high as $75.88 per share
and as low as $1.41 per share. Between January 1, 2007 and December 31, 2009,
the price has ranged between a high of $9.85 per share and a low of $1.95 per
share. The significant market price fluctuations of our common stock are due to
a variety of factors, including:
- the demand in the market for our
common stock;
- the experimental nature of our
product candidates;
- fluctuations in our operating
results;
- market conditions relating to the
biopharmaceutical and pharmaceutical industries;
- announcements of technological
innovations, new commercial products, or clinical progress or lack thereof by us, our collaborative
partners or our competitors;
- announcements concerning
regulatory developments, developments with respect to proprietary rights and our
collaborations;
- comments by securities
analysts;
- general market
conditions;
- political developments related to
hESC research;
- public concern with respect to our
product candidates; or
- the issuance of common stock to partners, vendors or to investors to raise additional capital.
In addition, the stock market is subject to other factors outside our
control that can cause extreme price and volume fluctuations. In the third and
fourth quarters of 2008, as well as during 2009, broad distress in the financial
markets and the economy have resulted in greatly increased market uncertainty
and instability in both U.S. and international capital and credit markets. These
conditions, combined with volatile oil prices, declining business and consumer
confidence and increased unemployment have recently contributed to substantial
market volatility, and if such market conditions persist, the price of our
common stock may fluctuate or decline. Securities class action litigation has
often been brought against companies, including many biotechnology companies,
which experience volatility in the market price of their securities. Litigation
brought against us could result in substantial costs and a diversion of
management’s attention and resources, which could adversely affect our
business.
The sale of a substantial number of shares may adversely affect the
market price for our common stock.
The sale of a substantial number of shares of our common stock in the
public market, or the perception that such sales could occur, could
significantly and negatively affect the market price for our common stock. As of
December 31, 2009, we had 200,000,000 shares of common stock authorized for
issuance and 92,521,946 shares of common stock outstanding. In addition, as of
December 31, 2009, we have reserved for future issuance approximately 25,070,022
shares of common stock for our stock plans, potential milestone payments and
outstanding warrants.
In addition, we have issued common stock to certain parties, such as
vendors and service providers, as payment for products and services. Under these
arrangements, we typically agree to register the shares for resale soon after
their issuance. We may continue to pay for certain goods and services in this
manner, which would dilute your interest in us. Also, sales of the shares issued
in this manner could negatively affect the market price for our common
stock.
32
Our undesignated preferred stock may inhibit potential acquisition bids;
this may adversely affect the market price for our common stock and the voting
rights of holders of our common stock.
Our certificate of incorporation provides our Board of Directors with the
authority to issue up to 3,000,000 shares of undesignated preferred stock and to
determine or alter the rights, preferences, privileges and restrictions granted
to or imported upon these shares without further vote or action by our
stockholders. As of the date of this Form 10-K, 50,000 shares of preferred stock
have been designated Series A Junior Participating Preferred Stock and the Board
of Directors still has authority to designate and issue up to 2,950,000 shares
of preferred stock in one or more classes or series. The issuance of shares of
preferred stock may delay or prevent a change in control transaction without
further action by our stockholders. As a result, the market price for our common
stock may be adversely affected.
In addition, if we issue preferred stock in the future that has
preference over our common stock with respect to the payment of dividends or
upon our liquidation, dissolution or winding up, or if we issue preferred stock
with voting rights that dilute the voting power of our common stock, the rights
of holders of our common stock or the market price for our common stock could be
adversely affected.
Provisions in our share purchase rights plan, charter and bylaws, and
provisions of Delaware law, may inhibit potential acquisition bids for us, which
may prevent holders of our common stock from benefiting from what they believe
may be the positive aspects of acquisitions and
takeovers.
Our Board of Directors has adopted a share purchase rights plan, commonly
referred to as a “poison pill.” This plan entitles existing stockholders to
rights, including the right to purchase shares of common stock, in the event of
an acquisition of 15% or more of our outstanding common stock.
Our share purchase rights plan could prevent stockholders from profiting
from an increase in the market value of their shares as a result of a change of
control of us by delaying or preventing a change of control. In addition, our
Board of Directors has the authority, without further action by our
stockholders, to issue additional shares of common stock, and to fix the rights
and preferences of one or more series of preferred stock.
In addition to our share purchase rights plan and the undesignated
preferred stock, provisions of our charter documents and bylaws may make it
substantially more difficult for a third party to acquire control of us and may
prevent changes in our management, including provisions that:
- prevent stockholders from taking
actions by written consent;
- divide the Board of Directors into
separate classes with terms of office that are structured to prevent all of the directors from being
elected in any one year; and
- set forth procedures for nominating directors and submitting proposals for consideration at stockholders’ meetings.
Provisions of Delaware law may also inhibit potential acquisition bids
for us or prevent us from engaging in business combinations. In addition, we
have severance agreements with several employees and a change of control
severance plan which could require an acquiror to pay a higher price. Either
collectively or individually, these provisions may prevent holders of our common
stock from benefiting from what they may believe are the positive aspects of
acquisitions and takeovers, including the potential realization of a higher rate
of return on their investment from these types of transactions.
We do not intend to pay cash dividends on our common stock in the
foreseeable future.
We do not anticipate paying cash dividends on our common stock in the
foreseeable future. Any payment of cash dividends will depend upon our financial
condition, results of operations, capital requirements and other factors and
will be at the discretion of the Board of Directors. Furthermore, we may incur
additional indebtedness that may severely restrict or prohibit the payment of
dividends.
33
Failure to achieve and maintain effective internal controls in accordance
with Section 404 of the Sarbanes-Oxley Act of 2002 could have a material adverse
effect on our business and stock price.
Section 404 of the Sarbanes-Oxley Act of 2002 (the Sarbanes-Oxley Act)
requires that we establish and maintain an adequate internal control structure
and procedures for financial reporting. Our annual report on Form 10-K must
contain an assessment by management of the effectiveness of our internal control
over financial reporting and must include disclosure of any material weaknesses
in internal control over financial reporting that we have identified. In
addition, our independent registered public accounting firm must annually
provide an opinion on the effectiveness of our internal control over financial
reporting.
The requirements of Section 404 of the Sarbanes-Oxley Act are ongoing and
also apply to future years. We expect that our internal control over financial
reporting will continue to evolve as our business develops. Although we are
committed to continue to improve our internal control processes and we will
continue to diligently and vigorously review our internal control over financial
reporting in order to ensure compliance with Section 404 requirements, any
control system, regardless of how well designed, operated and evaluated, can
provide only reasonable, not absolute, assurance that its objectives will be
met. Therefore, we cannot be certain that in the future material weaknesses or
significant deficiencies will not exist or otherwise be discovered. If material
weaknesses or other significant deficiencies occur, these weaknesses or
deficiencies could result in misstatements of our results of operations,
restatements of our consolidated financial statements, a decline in our stock
price, or other material adverse effects on our business, reputation, results of
operations, financial condition or liquidity.
ITEM 1B. UNRESOLVED STAFF
COMMENTS
None.
ITEM 2. PROPERTIES
We currently lease approximately 41,000 square feet of office space at
200 and 230 Constitution Drive, Menlo Park, California. The leases for 200 and
230 Constitution Drive expire in July 2012. We have an option to extend the
leases for one additional period of four years. In March 2008, as payment of the
total rent due for the premises at 200 and 230 Constitution Drive, we issued
742,158 shares of our common stock to the lessor of those premises. As a result,
we have no cash rental obligation from August 1, 2008 through July 31, 2012. We
also currently lease approximately 14,500 square feet of office space at 149
Commonwealth Drive, Menlo Park, California. The lease for 149 Commonwealth Drive
expires in April 2010. In May 2007, as payment of the total rent due for the
premises at 149 Commonwealth Drive, we issued 210,569 shares of our common stock
to the lessor of those premises. As a result, we have no cash rental obligation
from May 1, 2007 through April 30, 2010. In January 2010, we extended the lease
for the premises at 149 Commonwealth Drive to July 31, 2012. In connection with
that lease extension, we issued 94,741 shares of our common stock to the lessor
of those premises as a first installment payment of the total rent due under the
extended lease which pays for our cash rental obligation from May 1, 2010
through May 31, 2011. We believe that our existing facilities are adequate to
meet our requirements for the near term.
ITEM 3. LEGAL PROCEEDINGS
None.
ITEM 4. SUBMISSION OF MATTERS TO A VOTE OF
SECURITY HOLDERS
None.
34
PART II
| ITEM 5. | MARKET FOR THE REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our common stock is quoted on the Nasdaq Global Market under the symbol
GERN. The high and low closing sales prices as reported by the Nasdaq Global
Market of our common stock for each of the quarters in the years ended December
31, 2009 and 2008 are as follows:
| High | Low | ||||
| Year ended December 31, 2009 | |||||
| First quarter | $ | 8.15 | $ | 3.79 | |
| Second quarter | $ | 7.67 | $ | 4.41 | |
| Third quarter | $ | 9.17 | $ | 6.48 | |
| Fourth quarter | $ | 7.08 | $ | 5.19 | |
| Year ended December 31, 2008 | |||||
| First quarter | $ | 5.73 | $ | 4.04 | |
| Second quarter | $ | 5.40 | $ | 3.45 | |
| Third quarter | $ | 4.86 | $ | 3.17 | |
| Fourth quarter | $ | 4.67 | $ | 2.23 | |
As of February 23, 2010, there were approximately 796 stockholders of
record. We are engaged in a highly dynamic industry, which often results in
significant volatility of our common stock price. On February 23, 2010, the
closing sales price for our common stock was $5.72 per share.
Dividend Policy
We have never paid cash dividends on our capital stock and do not
anticipate paying cash dividends in the foreseeable future, but intend to retain
our capital resources for reinvestment in our business. Any future determination
to pay cash dividends will be at the discretion of the Board of Directors and
will be dependent upon our financial condition, results of operations, capital
requirements and other factors as the Board of Directors deems relevant.
Performance Measurement Comparison (1)
The following graph compares total stockholder returns of Geron
Corporation for the last five fiscal years beginning December 31, 2004 to two
indices: the Nasdaq CRSP Total Return Index for the Nasdaq Stock Market-U.S.
Companies (the Nasdaq-US) and the Nasdaq Pharmaceutical Index (the
Nasdaq-Pharmaceutical). The total return for our stock and for each index
assumes the reinvestment of dividends, although we have never declared dividends
on Geron stock, and is based on the returns of the component companies weighted
according to their capitalizations as of the end of each quarterly period. The
Nasdaq-US tracks the aggregate price performance of equity securities of U.S.
companies traded on the Nasdaq Global Market (NGM). The Nasdaq-Pharmaceutical,
which is calculated and supplied by Nasdaq, represents pharmaceutical companies,
including biotechnology companies, trading on Nasdaq under the Standard
Industrial Classification (SIC) Code No. 283 Drugs main category (2833 —
Medicinals & Botanicals, 2834 — Pharmaceutical Preparations, 2835 —
Diagnostic Substances, 2836 — Biological Products). Geron common stock trades on
the NGM and is a component of both the Nasdaq-US and the
Nasdaq-Pharmaceutical.
____________________
____________________
| (1) | This Section is not “soliciting material,” is not deemed “filed” with the SEC and is not to be incorporated by reference in any filing of the Company under the Securities Act, or the Exchange Act, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing. |
35
Comparison of Five Year Cumulative Total
Return on Investment Among
Geron Corporation, the Nasdaq-US Index and the Nasdaq-Pharmaceutical Index(2)
Geron Corporation, the Nasdaq-US Index and the Nasdaq-Pharmaceutical Index(2)
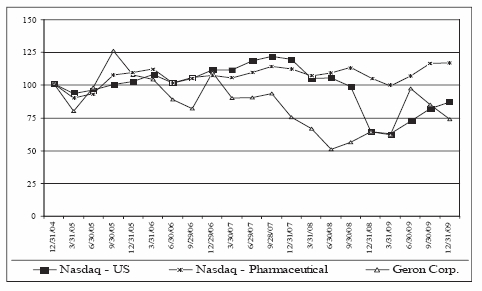
____________________
| (2) | Shows the cumulative total return on investment assuming an investment of $100 in each of Geron, the Nasdaq-US and the Nasdaq-Pharmaceutical on December 31, 2004. The cumulative total return on Geron stock has been computed based on a price of $7.97 per share, the price at which Geron’s shares closed on December 31, 2004. |
Recent Sales of Unregistered
Securities
On November 10, 2009, we issued 55,545 shares of our common stock to
Hongene Biotechnology Limited (Hongene) in a private placement as advance
consideration related to an addendum agreement to a manufacturing agreement
pursuant to which Hongene is making certain raw materials for us intended to be
used for the manufacture of drug product for use in human clinical trials. The
total fair value of the common stock was $304,000 which has been recorded as a
prepaid asset and is being amortized to research and development expense on a
pro-rata basis upon the proper receipt of materials which is expected to be
within six months. As of December 31, 2009, $304,000 remained as a prepaid
asset.
On November 10, 2009, we issued 93,244 shares of our common stock to
Samchully Pharm. Co., Ltd. (Samchully) in a private placement as advance
consideration related to an addendum agreement to a manufacturing agreement
pursuant to which Samchully is performing certain services and manufacturing
certain raw materials and products for us intended for use in human clinical
trials. The total fair value of the common stock was $511,000 which has been
recorded as a prepaid asset and is being amortized to research and development
expense on a pro-rata basis upon the performance of services and the proper
receipt of materials which is expected to be over six months. As of December 31,
2009, $461,000 remained as a prepaid asset.
On November 10, 2009, we issued 195,331 shares of our common stock to
ReSearch Pharmaceutical Services, Inc. (RPS) as advance consideration related to
a first project agreement to a master services agreement under which RPS is
providing certain services in support of our clinical programs. The total fair
value of the common stock was $1.1 million which has been recorded as a prepaid
asset and is being amortized to research and development expense on a pro-rata
basis as services are performed which is expected to be over two years. As of
December 31, 2009, $963,000 remained as a prepaid asset.
We issued the above-described shares of common stock in reliance upon the
exemption from registration provided by Section 4(2) of the Securities Act of
1933, as amended. Hongene, Samchully and RPS represented to us that they are
accredited investors as defined in Rule 501(a) of the Securities Act of 1933, as
amended, and that the securities issued pursuant thereto were being acquired for
investment purposes.
36
Securities Authorized for Issuance under
Equity Compensation Plans
The information required by this
Item concerning our equity compensation plans is incorporated by reference from
the section captioned “Equity Compensation Plans” contained in our Definitive
Proxy Statement related to the annual meeting of stockholders to be held May 19,
2010, to be filed with the Securities and Exchange Commission.
ITEM 6. SELECTED FINANCIAL
DATA
| Year Ended December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| (In thousands, except share and per share data) | ||||||||||||||||||||
| Consolidated Statements of | ||||||||||||||||||||
| Operations Data: | ||||||||||||||||||||
| Revenues from collaborative agreements | $ | 450 | $ | 294 | $ | 672 | $ | 622 | $ | 290 | ||||||||||
| License fees and royalties | 1,276 | 2,509 | 6,950 | 2,655 | 5,868 | |||||||||||||||
| Total revenues | 1,726 | 2,803 | 7,622 | 3,277 | 6,158 | |||||||||||||||
| Operating expenses: | ||||||||||||||||||||
| Research and development | 57,617 | 53,664 | 54,624 | 41,234 | 35,080 | |||||||||||||||
| General and administrative | 14,343 | 16,183 | 15,837 | 9,403 | 8,788 | |||||||||||||||
| Total operating expenses | 71,960 | 69,847 | 70,461 | 50,637 | 43,868 | |||||||||||||||
| Loss from operations | (70,234 | ) | (67,044 | ) | (62,839 | ) | (47,360 | ) | (37,710 | ) | ||||||||||
| Unrealized gain (loss) on fair value | ||||||||||||||||||||
| of derivatives | 157 | 418 | 15,453 | 7,421 | (161 | ) | ||||||||||||||
| Interest and other income | 1,374 | 5,542 | 10,791 | 8,704 | 4,658 | |||||||||||||||
| Equity in losses of joint venture | — | — | — | — | (12 | ) | ||||||||||||||
| Losses recognized under equity | ||||||||||||||||||||
| method investment | (1,338 | ) | (844 | ) | — | — | — | |||||||||||||
| Interest and other expense | (143 | ) | (93 | ) | (102 | ) | (130 | ) | (464 | ) | ||||||||||
| Net loss | (70,184 | ) | (62,021 | ) | (36,697 | ) | (31,365 | ) | (33,689 | ) | ||||||||||
| Deemed dividend on derivatives (1) | (190 | ) | — | (9,081 | ) | — | — | |||||||||||||
| Net loss applicable to common | ||||||||||||||||||||
| stockholders | $ | (70,374 | ) | $ | (62,021 | ) | $ | (45,778 | ) | $ | (31,365 | ) | $ | (33,689 | ) | |||||
| Basic and diluted net loss per share: | ||||||||||||||||||||
| Net loss per share applicable to common | ||||||||||||||||||||
| stockholders | $ | (0.80 | ) | $ | (0.79 | ) | $ | (0.62 | ) | $ | (0.47 | ) | $ | (0.58 | ) | |||||
| Shares used in computing net loss per share | ||||||||||||||||||||
| applicable to common stockholders | 88,078,557 | 78,187,795 | 74,206,249 | 66,057,367 | 57,879,725 | |||||||||||||||
| (1) | In April 2009 in connection with our continued collaboration with an investor and licensee and the data received under the collaboration relevant to Geron’s therapeutic programs, we modified the terms of certain outstanding warrants held by this investor by extending the exercise term and reducing the exercise price. The exercise term of warrants to purchase 200,000 shares of common stock was extended to March 9, 2012 from March 9, 2010 and the exercise price was modified to $17.50 per share from $67.09 per share. The exercise term of warrants to purchase 100,000 shares of common stock was extended to March 9, 2012 from March 9, 2010 and the exercise price was unchanged at $12.50 per share. In connection with the modifications, we recognized a deemed dividend of approximately $190,000 in our consolidated statements of operations for the incremental fair value of the modified warrants. | |
| In February 2007 in exchange for the exercise of certain warrants, we issued new warrants to the same institutional investors. The aggregate fair value of $3.7 million for the new warrants was recognized as a deemed dividend. In December 2007, we modified the terms of certain outstanding warrants by extending the exercise term and reducing the exercise price. In connection with the | ||
37
modifications, we received $3.6 million in cash consideration from the
institutional investors holding the outstanding warrants. We recognized a deemed
dividend of $5.4 million for the incremental fair value of the modified
warrants, net of the cash consideration received from the institutional
investors for the modifications.
| December 31, | ||||||||||||||||||||
| 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Consolidated Balance Sheet Data: | ||||||||||||||||||||
| Cash, restricted cash, cash equivalents and | ||||||||||||||||||||
| marketable securities | $ | 167,070 | $ | 163,655 | $ | 208,444 | $ | 213,860 | $ | 191,003 | ||||||||||
| Working capital | 110,324 | 160,535 | 200,655 | 170,377 | 171,310 | |||||||||||||||
| Total assets | 180,382 | 176,218 | 218,896 | 220,800 | 201,243 | |||||||||||||||
| Long-term obligations | — | — | 427 | — | — | |||||||||||||||
| Accumulated deficit | (577,267 | ) | (506,893 | ) | (444,872 | ) | (399,094 | ) | (367,729 | ) | ||||||||||
| Total stockholders’ equity | 172,577 | 168,455 | 205,674 | 173,919 | 175,698 | |||||||||||||||
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS
OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Overview
The following discussion should be
read in conjunction with the audited consolidated financial statements and notes
thereto included in Part II, Item 8 of this annual report.
Geron is developing first-in-class
biopharmaceuticals for the treatment of cancer and chronic degenerative
diseases, including spinal cord injury, heart failure and diabetes. The company
is advancing an anti-cancer drug and a cancer vaccine that target the enzyme
telomerase through multiple clinical trials in different cancers.
Critical Accounting Policies and Estimates
Our consolidated financial
statements have been prepared in accordance with accounting principles generally
accepted in the United States of America. The preparation of these financial
statements requires management to make estimates and assumptions that affect the
reported amounts of assets, liabilities, revenues and expenses. Note 1 of Notes
to Consolidated Financial Statements describes the significant accounting
policies used in the preparation of the consolidated financial statements.
Certain of these significant accounting policies are considered to be critical
accounting policies, as defined below.
A critical accounting policy is
defined as one that is both material to the presentation of our financial
statements and requires management to make difficult, subjective or complex
judgments that could have a material effect on our financial condition and
results of operations. Specifically, critical accounting estimates have the
following attributes: (i) we are required to make assumptions about matters that
are highly uncertain at the time of the estimate; and (ii) different estimates
we could reasonably have used, or changes in the estimate that are reasonably
likely to occur, would have a material effect on our financial condition or
results of operations.
Estimates and assumptions about
future events and their effects cannot be determined with certainty. We base our
estimates on historical experience and on various other assumptions believed to
be applicable and reasonable under the circumstances. These estimates may change
as new events occur, as additional information is obtained and as our operating
environment changes. These changes have historically been minor and have been
included in the consolidated financial statements as soon as they became known.
Based on a critical assessment of our accounting policies and the underlying
judgments and uncertainties affecting the application of those policies,
management believes that our consolidated financial statements are fairly stated
in accordance with accounting principles generally accepted in the United
States, and meaningfully present our financial condition and results of
operations.
38
We believe the following critical accounting policies reflect our more
significant estimates and assumptions used in the preparation of our
consolidated financial statements:
Revenue Recognition
Since our inception, a substantial portion of our revenues has been
generated from research and licensing agreements. Revenue under such agreements
typically includes upfront signing or license fees, cost reimbursements,
milestone payments and royalties on future product sales.
We recognize nonrefundable signing, license or non-exclusive option fees
as revenue when rights to use the intellectual property related to the license
have been delivered and over the term of the agreement if we have continuing
performance obligations. We recognize milestone payments, which are subject to
substantive contingencies, upon completion of specified milestones, which
represents the culmination of an earnings process, according to contract terms.
Royalties are generally recognized as revenue upon the receipt of the related
royalty payment. We recognize cost reimbursement revenue under collaborative
agreements as the related research and development costs for services are
rendered. We recognize related party revenue under collaborative agreements as
the related party research and development costs for services are rendered and
when the source of funds have not been derived from our contributions to the
related party. Deferred revenue represents the portion of research or license
payments received which have not been earned. When payments are received in
equity securities, we do not recognize any revenue unless such securities are
determined to be realizable in cash.
We estimate the projected future term of license agreements over which we
recognize revenue. Our estimates are based on contractual terms, historical
experience and general industry practice. Revisions in the estimated terms of
these license agreements have the effect of increasing or decreasing license fee
revenue in the period of revision. As of December 31, 2009, no revisions to the
estimated future terms of license agreements have been made and we do not expect
revisions to the currently active agreements in the future.
Valuation of Stock-Based Compensation
We measure and recognize compensation expense for all stock-based awards
to our employees and directors, including employee stock options, restricted
stock awards and employee stock purchases related to our Employee Stock Purchase
Plan (ESPP) based on estimated fair values. We estimate the fair value of
stock-based awards using the Black Scholes option-pricing model. Option-pricing
model assumptions such as expected volatility, risk-free interest rate and
expected term impact the fair value estimate. Further, the estimated forfeiture
rate impacts the amount of aggregate compensation recognized during the period.
The fair value of stock-based awards is amortized over the vesting period of the
awards using a straight-line method.
Expected volatilities are based on historical volatilities of our stock
since traded options on Geron stock do not correspond to option terms and
trading volume of options is limited. The expected term of options represents
the period of time that options granted are expected to be outstanding. In
deriving this assumption, we reviewed actual historical exercise and
cancellation data and the remaining outstanding options not yet exercised or
cancelled. The expected term of employees’ purchase rights, under our ESPP, is
equal to the purchase period. The risk-free interest rate is based on the U.S.
Zero Coupon Treasury Strip Yields for the expected term in effect on the date of
grant. Forfeiture rate was estimated based on historical experience and will be
adjusted over the requisite service period based on the extent to which actual
forfeitures differ, or are expected to differ, from their estimate.
We annually evaluate the assumptions used in estimating fair values of
our stock-based awards by reviewing current trends in comparison to historical
data. We have not revised the method in which we derive assumptions in order to
estimate fair values of our stock-based awards. If factors change and we employ
different assumptions in future periods, the stock-based compensation expense
that we record for awards to employees and directors may differ significantly
from what we have recorded in the current period.
39
Non-cash compensation expense recognized for stock-based awards to
employees and directors was $10.6 million, $11.5 million and $11.4 million for
the years ended December 31, 2009, 2008 and 2007, respectively. As of December
31, 2009, total non-cash compensation cost related to unvested stock-based
awards not yet recognized was $17.6 million, net of estimated forfeitures, which
is expected to be recognized over the next 41 months on a weighted-average
basis.
For our non-employee stock-based awards, the measurement date on which
the fair value of the stock-based award is calculated is equal to the earlier of
(i) the date at which a commitment for performance by the counterparty to earn
the equity instrument is reached or (ii) the date at which the counterparty’s
performance is complete. We recognized non-cash stock-based compensation expense
of $190,000, zero and $1.5 million for the fair value of the vested portion of
non-employee options, restricted stock awards and warrants in our consolidated
statements of operations for 2009, 2008 and 2007, respectively.
Fair Value of Financial Instruments
We categorize assets and liabilities recorded at fair value on our
consolidated balance sheet based upon the level of judgment associated with
inputs used to measure their fair value. The categories are as follows:
Level 1 – Inputs are unadjusted, quoted prices in active markets for
identical assets or liabilities at the measurement date. An active market for
the asset or liability is a market in which transactions for the asset or
liability occur with sufficient frequency and volume to provide pricing
information on an ongoing basis.
Level 2 – Inputs (other than quoted market prices included in Level 1)
are either directly or indirectly observable for the asset or liability through
correlation with market data at the measurement date and for the duration of the
instrument’s anticipated life.
Level 3 – Inputs reflect management’s best estimate of what market
participants would use in pricing the asset or liability at the measurement
date. Consideration is given to the risk inherent in the valuation technique and
the risk inherent in the inputs to the model.
A financial instrument’s categorization is based upon the lowest level of
input that is significant to the fair value measurement. Following is a
description of the valuation methodologies used for instruments measured at fair
value on our consolidated balance sheet, including the category for such
instruments.
We classify inputs to derive fair values for marketable debt securities
available-for-sale and marketable investments in licensees as Level 1 and 2.
Instruments classified as Level 1 include U.S. Treasury securities, U.S.
government-sponsored enterprise securities, money market funds and publicly
traded equity securities in active markets, representing 64% of total financial
assets measured at fair value as of December 31, 2009. Instruments classified as
Level 2 include corporate notes, representing 36% of total financial assets
measured at fair value as of December 31, 2009. The price for each security at
the measurement date is derived from various sources. Periodically, we assess
the reasonableness of these sourced prices by comparing them to the prices
provided by our portfolio managers from broker quotes. Historically, we have not
experienced significant deviation between the sourced prices and our portfolio
manager’s prices.
Warrants to purchase common stock and non-employee options are normally
traded less actively, have trade activity that is one way, and/or traded in
less-developed markets and are therefore valued based upon models with
significant unobservable market parameters, resulting in Level 3 categorization.
The fair value for these instruments is calculated using the Black Scholes
option-pricing model. The model’s inputs reflect assumptions that market
participants would use in pricing the instrument in a current period
transaction. Inputs to the model include stock volatility, dividend yields,
expected term of the derivatives and risk-free interest rates. See the following
discussion, “Fair Value of Derivatives,” for information on derivation of inputs
to the model. Changes to the model’s inputs are not changes to valuation
methodologies, but instead reflect direct or indirect impacts from changes in
market conditions. Accordingly, results from the valuation model in one period
may not be indicative of future period measurements. Instruments classified as
Level 3 include derivative liabilities, representing all of total financial
liabilities measured at fair value as of December 31, 2009.
40
For a further discussion regarding fair value measurements, see Note 2 on
Fair Value Measurements of Notes to Consolidated Financial
Statements.
Fair Value of Derivatives
For warrants and non-employee options classified as assets or
liabilities, the fair value of these instruments is recorded on the consolidated
balance sheet at inception of such classification and marked to fair value at
each financial reporting date. The change in fair value of the warrants and
non-employee options is recorded in the consolidated statements of operations as
an unrealized gain (loss) on fair value of derivatives. The warrants and
non-employee options continue to be reported as an asset or liability until such
time as the instruments are exercised or expire or are otherwise modified to
remove the provisions which require this treatment, at which time these
instruments are marked to fair value and reclassified from assets or liabilities
to stockholders’ equity. For warrants and non-employee options classified as
permanent equity, the fair value of the warrants and non-employee options is
recorded in stockholders’ equity and no further adjustments are made.
Fair value of warrants and non-employee options is estimated using the
Black Scholes option-pricing model. Use of this model requires us to make
assumptions regarding stock volatility, dividend yields, expected term of the
warrants and non-employee options and risk-free interest rates. Expected
volatilities are based on historical volatilities of our stock. The expected
term of warrants and non-employee options represent the remaining contractual
term of the instruments. The risk-free interest rate is based on the U.S. Zero
Coupon Treasury Strip Yields for the remaining term of the instrument. If
factors change and we employ different assumptions in future periods, the fair
value of these warrants and non-employee options reflected as of each balance
sheet date and the resulting change in fair value that we record may differ
significantly from what we have recorded in previous periods. As of December 31,
2009, we have not revised the method in which we derive assumptions in order to
estimate fair values of warrants and non-employee options classified as assets
or liabilities, and we do not expect revisions in the future.
Results of Operations
Our results of operations have fluctuated from period to period and may
continue to fluctuate in the future, based upon the progress of our research and
development efforts and variations in the level of expenses related to
developmental efforts during any given period. Results of operations for any
period may be unrelated to results of operations for any other period. In
addition, historical results should not be viewed as indicative of future
operating results. We are subject to risks common to companies in our industry
and at our stage of development, including risks inherent in our research and
development efforts, reliance upon our collaborative partners, enforcement of
our patent and proprietary rights, need for future capital, potential
competition and uncertainty of preclinical and clinical trial results or
regulatory approvals or clearances. In order for a product candidate to be
commercialized based on our research, we and our collaborators must conduct
preclinical tests and clinical trials, demonstrate the efficacy and safety of
our product candidates, obtain regulatory approvals or clearances and enter into
manufacturing, distribution and marketing arrangements, as well as obtain market
acceptance. We do not expect to receive revenues or royalties based on
therapeutic products for a period of years, if at all.
Revenues
We recognized $450,000 of revenues from collaborative agreements in 2009
compared to $294,000 in 2008 and $672,000 in 2007. Revenues in 2009 primarily
reflected revenue recognized under our collaboration with GE Healthcare, Ltd.
(GE Healthcare). Revenues in 2008 and 2007 primarily reflected related party
reimbursements we received from our joint venture in Hong Kong, TA Therapeutics,
Ltd. (TAT), for scientific research services and revenue recognized under our
collaboration with Corning Life Sciences. Since June 16, 2007, we have been
including TAT’s results in our consolidated financial statements and have
eliminated any related party revenue when the source of funds has been derived
from our contributions to the related party. Prior to that date, related party
revenue earned under the contract to perform scientific research services for
TAT was recognized as revenue as the services were performed.
We have entered into license and option agreements with companies
involved with oncology, diagnostics, research tools, agriculture and biologics
production. In each of these agreements, we have granted certain rights to our
technologies. In connection with the agreements, we are entitled to receive
license fees, option
41
fees, milestone payments
and royalties on future sales, or any combination thereof. We recognized license
fee revenues of $1.1 million, $2.1 million and $6.7 million in 2009, 2008 and
2007, respectively, related to our various agreements. License fee revenue in
2009 primarily reflected revenue recognized from upfront license fee payments
under our collaboration with GE Healthcare. License fee revenue in 2008
primarily reflected the receipt of a $1.5 million milestone payment from Exeter
Life Sciences, Inc. as a result of the final Risk Assessment released by the
U.S. Food and Drug Administration (FDA) addressing food products made from
cloned animals or their progeny. License fee revenue in 2007 primarily reflected
the receipt of $5.0 million in milestone payments in connection with the
collaboration and license agreement with Merck & Co., Inc. (Merck). We
expect to recognize revenue of $700,000 in 2010 and $350,000 in 2011 related to
our existing deferred revenue. Current revenues may not be predictive of future
revenues.
We recognized royalty revenues of $160,000, $403,000 and $211,000 in
2009, 2008 and 2007, respectively, on product sales of telomerase detection and
telomere measurement kits to the research-use-only market, telomerase-based
research products and agricultural products. License and royalty revenues are
dependent upon additional agreements being signed and future product
sales.
Research and Development Expenses
Research and development expenses were $57.6 million, $53.7 million and
$54.6 million for the years ended December 31, 2009, 2008 and 2007,
respectively. The increase in 2009 compared to 2008 was primarily the result of
higher personnel related costs of $2.3 million in connection with additional
clinical operations personnel and increased clinical trial costs of $846,000 as
a result of increased patient enrollment for imetelstat and GRNVAC1 trials. The
decrease in 2008 compared to 2007 was primarily the net result of decreased
manufacturing costs of $1.1 million as a result of timing of drug purchases for
imetelstat and lower scientific supplies of $1.6 million, partially offset by
increased clinical trial costs of $2.0 million associated with imetelstat and
GRNVAC1. Overall, we expect research and development expenses to increase as we
incur expenses related to clinical trials for imetelstat along with continued
development of our human embryonic stem cell (hESC) programs.
Our research and development activities have arisen from our two major
technology platforms, telomerase and hESCs. The oncology programs focus on
treating or diagnosing cancer by targeting or detecting the presence of
telomerase, either inhibiting activity of the telomerase enzyme, diagnosing
cancer by detecting the presence of telomerase, or using telomerase as a target
for therapeutic vaccines. Our core knowledge base in telomerase and telomere
biology supports all these approaches, and our scientists may contribute to any
or all of these programs in a given period. The following table briefly
describes our cancer therapeutic product candidates and their stage of
development:
| Patient | ||||
| Product | Development | Enrollment | ||
| Product | Description | Application | Stage | Status |
| Imetelstat | Telomerase | Chronic Lymphoproliferative | Phase I Trial | Completed |
| (GRN163L) | Inhibitor | Diseases | (single agent) | |
| Imetelstat | Telomerase | Solid Tumors | Phase I Trial | Open |
| (GRN163L) | Inhibitor | (single agent) | ||
| Imetelstat | Telomerase | Multiple Myeloma* | Phase I Trial | Completed |
| (GRN163L) | Inhibitor | (single agent) | ||
| Imetelstat | Telomerase | Non-Small Cell Lung | Phase I Trial | Completed |
| (GRN163L) | Inhibitor | Cancer* | (combination) | |
| Imetelstat | Telomerase | Breast Cancer* | Phase I/II Trial | Open |
| (GRN163L) | Inhibitor | (combination) | ||
| Imetelstat | Telomerase | Multiple Myeloma | Phase I Trial | Completed |
| (GRN163L) | Inhibitor | (combination) | ||
| GRNVAC1 | Telomerase | Acute Myelogenous | Phase II Trial | Completed |
| Cancer Vaccine | Leukemia (AML) | |||
| * Initiation of Phase II clinical trials in multiple myeloma, non-small cell lung cancer, breast cancer and essential thrombocythemia is planned for 2010. | ||||
42
Interim data from the Phase I single agent trial in patients with
relapsed and refractory multiple myeloma has shown that imetelstat inhibits
telomerase both in the bulk myeloma fraction as well as the stem-cell containing
fraction in patients’ bone marrow. Interim data from the trial in patients with
refractory, advanced solid cancers has shown that with a modified dosing
schedule, the exposures to imetelstat exceeded the levels associated with
inhibiting tumor growth from several models of human cancers. From the above
trials, we have obtained data to assess the safety, tolerability,
pharmacokinetics and pharmacodynamics of imetelstat. With this information, we
have established the single agent Phase II dose and dosing schedule and are
planning to advance imetelstat to Phase II clinical trials in four different
malignancies in 2010.
Taking the results from the Duke University clinical studies in prostate
cancer, hematologic malignancies and renal cell carcinoma, we optimized the
vaccine manufacturing process and transferred it to a contract manufacturer. We
are conducting a Phase II clinical trial of our telomerase vaccine using the
prime/boost vaccination protocol in patients with acute myelogenous leukemia in
complete clinical remission. Twenty patients in the study have received
vaccination with GRNVAC1. Recent data from the trial showed that GRNVAC1 was
safe and generally well tolerated over multiple vaccinations. Of the 20 patients
in the study, 14 remain in complete clinical remission. Positive overall immune
responses were detected in 12 out of 20 patients. No correlation has been made
between positive immune response and patient remission status. Continued
follow-up of patients for an additional nine months is required to estimate the
impact of vaccination on disease-free survival.
Our hESC therapy programs focus on treating injuries and degenerative
diseases with cell therapies based on cells derived from hESCs. A core of
knowledge of hESC biology, as well as a significant continuing effort in
deriving, growing, maintaining, and differentiating hESCs, underlies all aspects
of this group of programs. Many of our researchers are allocated to more than
one hESC program, and the percentage allocations of time change as the resource
needs of individual programs vary. In our hESC therapy programs, we have
concentrated our resources on several specific cell types,
including:
- GRNOPC1, hESC-derived
oligodendrocyte progenitor cells, for the treatment of acute spinal
cord injury;
- GRNCM1, hESC-derived
cardiomyocytes, for toxicology drug testing and the treatment of myocardial disease;
- GRNIC1, hESC-derived pancreatic
islet ß cells for the treatment of diabetes;
- hESC-derived chondrocytes for the
treatment of osteoarthritis;
- hESC-derived hepatocytes for ADME
drug testing;
- hESC-derived dendritic cells for
cancer immunotherapy and to prevent immune rejection of the other cell types used in therapeutic
applications; and
- hESC-derived osteoblasts for the treatment of osteoporosis.
We have developed proprietary methods to grow, maintain, and scale the
culture of undifferentiated hESCs that use feeder cell-free and serum-free media
with chemically defined components. We have also developed scalable processes to
differentiate these cells into therapeutically relevant cells. Currently, the
human clinical trial of GRNOPC1, our hESC-derived therapy targeted for the
treatment of acute spinal cord injury, has been placed on clinical hold by the
FDA. We are in discussions with the agency to answer its questions and proceed
with the clinical trial.
Research and development expenses
incurred under each of these programs are as follows (in thousands):
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Oncology | $ | 29,543 | $ | 30,259 | $ | 29,916 | |||
| hESC Therapies | 28,074 | 23,405 | 24,708 | ||||||
| Total | $ | 57,617 | $ | 53,664 | $ | 54,624 | |||
43
At this time, we cannot provide reliable estimates of how much time or
investment will be necessary to commercialize products from the programs
currently in progress. Drug development in the United States is a process that
includes multiple steps defined by the FDA under applicable statutes,
regulations and guidance documents. After the preclinical research process of
identifying, selecting and testing in animals a potential pharmaceutical
compound, the clinical development process begins with the filing of an
Investigational New Drug (IND) application. Clinical development typically
involves three phases of study: Phase I, II and III. The most significant costs
associated with clinical development are incurred in Phase III trials, which
tend to be the longest and largest studies conducted during the drug development
process. After the completion of a successful preclinical and clinical
development program, a New Drug Application (NDA) or Biologics License
Application (BLA) must be filed with the FDA, which includes, among other
things, very large amounts of preclinical and clinical data and results and
manufacturing-related information necessary to support requested approval of the
product. The NDA/BLA must be reviewed and approved by the FDA.
According to industry statistics, it generally takes 10 to 15 years to
research, develop and bring to market a new prescription medicine in the United
States. In light of the steps and complexities involved, the successful
development of our potential products is highly uncertain. Actual timelines and
costs to develop and commercialize a product are subject to enormous variability
and are very difficult to predict. In addition, various statutes and regulations
also govern or influence the manufacturing, safety reporting, labeling, storage,
record keeping and marketing of each product.
The lengthy process of seeking these regulatory reviews and approvals,
and the subsequent compliance with applicable statutes and regulations, require
the expenditure of substantial resources. Any failure by us to obtain, or any
delay in obtaining, regulatory approvals could materially adversely affect our
business. In responding to an NDA/BLA submission, the FDA may grant marketing
approval, may request additional information, may deny the application if it
determines that the application does not provide an adequate basis for approval,
and may also refuse to review an application that has been submitted if it
determines that the application does not provide an adequate basis for filing
and review. We cannot provide assurance that any approval required by the FDA
will be obtained on a timely basis, if at all.
For a more complete discussion of the risks and uncertainties associated
with completing development of potential products, see the sub-section titled
“Delays in the commencement of clinical testing of our current and potential
product candidates could result in increased costs to us and delay our ability
to generate revenues” and “Obtaining regulatory approvals to market our product
candidates in the United States and other countries is a costly and lengthy
process and we cannot predict whether or when we will be permitted to
commercialize our product candidates” in Part I, Item 1A entitled “Risk Factors”
and elsewhere in this annual report.
General and Administrative Expenses
General and administrative expenses were $14.3 million, $16.2 million and
$15.8 million for the years ended December 31, 2009, 2008 and 2007,
respectively. The decrease in 2009 from 2008 was primarily due to reduced legal
costs associated with our patents and lower audit fees and consulting costs. The
increase in 2008 from 2007 was primarily due to increased compensation expense
related to stock options and restricted stock awards to employees, partially
offset by reduced consulting expense and lower audit fees.
Unrealized Gain (Loss) on Fair Value of
Derivatives
Unrealized gain (loss) on fair value of derivatives reflects a non-cash
adjustment for changes in fair value of warrants to purchase common stock and
options held by non-employees that are classified as current liabilities.
Derivatives classified as assets or liabilities are marked to fair value at each
financial reporting date with any resulting unrealized gain (loss) recorded in
the consolidated statements of operations. The derivatives continue to be
reported as an asset or liability until such time as the instruments are
exercised or expire or are otherwise modified to remove the provisions which
require them to be recorded as assets or liabilities, at which time these
instruments are marked to fair value and reclassified from assets or liabilities
to stockholders’ equity. We incurred unrealized gains of $157,000, $418,000 and
$15.5 million for the years ended December 31, 2009, 2008 and 2007,
respectively.
44
Unrealized gains in 2009
and 2008 were due to the reduced fair values of derivatives resulting from
shortening of their contractual terms, decreases in the market value of our
stock and changes in other inputs factored into the estimate of their fair value
such as the volatility of our stock. Unrealized gains for 2007 were primarily
the result of amendments executed in March 2007 to certain warrant agreements to
address the presumption of net-cash settlement in the event that registered
shares were not available to settle the warrants enabling reclassification of
the decreasing fair value of those warrants from current liabilities to
stockholders’ equity. See Note 2 on Fair Value Measurements of Notes to
Consolidated Financial Statements of this Form 10-K for further discussion of
the fair value of derivatives.
Interest and Other Income
Interest income was $1.4 million, $5.5 million and $10.9 million for the
years ended December 31, 2009, 2008 and 2007, respectively. The decrease in 2009
compared to 2008 was primarily due to decreased interest rates. The decrease in
2008 compared to 2007 was primarily due to decreased interest rates and lower
cash and investment balances. Interest earned in future periods will depend on
the size of our securities portfolio and prevailing interest rates.
In 2009, net realized losses of $26,000 and in 2007, net realized gains
of $1,000 from sales of investments in licensees have been included in interest
and other income. No sales of investments in licensees occurred in 2008. Also
included in interest and other income for the years ended December 31, 2009,
2008 and 2007, were recognized losses of zero, $43,000 and $106,000,
respectively, related to other-than-temporary declines in fair value of our
investments in licensees.
Losses Recognized Under Equity Method
Investment
In August 2008, we exchanged our equity interest in the Start Licensing,
Inc. (Start) joint venture for equity interest in ViaGen, Inc. (ViaGen). In
September 2008, we provided a loan of $1.5 million to ViaGen in connection with
ViaGen’s acquisition of an interest in an unrelated company. The proceeds of the
loan did not fund prior ViaGen losses and represented additional financial
support to ViaGen.
In September 2009, we provided $3.6 million as a new equity investment in
ViaGen and also received $1.6 million from ViaGen in repayment of our loan,
resulting in a net investment of $2.0 million. The new investment in 2009 did
not fund prior ViaGen losses and represented additional financial support to
ViaGen. In accordance with the equity method of accounting, we recognized losses
of $1.3 million and $844,000 for 2009 and 2008, respectively, for our
proportionate share of ViaGen’s losses since providing the loan in September
2008. Previously, we had suspended the equity method of accounting for Start and
ViaGen since our proportionate share of net losses exceeded the value of our
investment and we had no commitments to provide financial support to either
company.
Interest and Other Expense
Interest and other expense was $143,000, $93,000 and $102,000 for the
years ended December 31, 2009, 2008 and 2007, respectively. In 2009, 2008 and
2007, interest and other expense was primarily comprised of bank charges. The
increase in interest and other expense for 2009 compared to 2008 was primarily
due to higher bank charges. The decrease in interest and other expense for 2008
compared to 2007 was primarily due to reduced bank charges as a result of lower
cash and investment balances.
Deemed Dividend on Derivatives
In April 2009, we modified the terms of certain outstanding warrants held
by an investor by extending the exercise term and, for certain of these
warrants, reducing the exercise price. In connection with the modifications, we
recognized a deemed dividend of approximately $190,000 in the consolidated
statements of operations for the incremental fair value of the modified
warrants, as calculated using the Black Scholes option-pricing model as of the
modification date.
45
In exchange for the exercise of warrants in February 2007, we issued
warrants to purchase 1,125,000 shares of common stock, at a premium, exercisable
from June 2007. The new warrants were substantially the same as the A Warrants
issued in connection with a financing in December 2006. The aggregate fair value
of $3.7 million for these new instruments, as calculated using the Black Scholes
option-pricing model, was recognized as a deemed dividend in the consolidated
statements of operations.
In December 2007, we modified the terms of certain outstanding warrants
by extending the exercise term and reducing the exercise price. The exercise
term of the 2004 A Warrants to purchase 2,295,082 shares of common stock was
extended to November 2011 and the exercise price was modified to $7.50 per
share. The exercise terms of the 2006 A Warrants to purchase 3,000,000 shares of
common stock and 2007 D Warrants to purchase 1,125,000 shares of common stock
were extended to December 2011 and the exercise prices were modified to $7.50
per share. In connection with the modifications, we received $3.6 million in
cash consideration from the institutional investors holding the outstanding
warrants. We recognized a deemed dividend of $5.4 million in the consolidated
statements of operations for the incremental fair value of the modified
warrants, as estimated using the Black Scholes option-pricing model as of the
modification date, net of the cash consideration received from the institutional
investors for the modifications.
Net Loss Applicable to Common Stockholders
Net loss applicable to common stockholders was $70.4 million, $62.0
million and $45.8 million for the years ended December 31, 2009, 2008 and 2007,
respectively. Overall net loss for 2009 increased compared to 2008 primarily due
to decreased interest income, reduced revenues from milestones, increased
research and development expenses and increased losses recognized for an equity
method investment. Overall net loss for 2008 increased compared to 2007
primarily due to reduced revenues from milestones, lower interest income and
decreased unrealized gains on derivatives.
Liquidity and Capital Resources
Cash, restricted cash, cash equivalents and marketable securities at
December 31, 2009 were $167.1 million, compared to $163.7 million at December
31, 2008 and $208.4 million at December 31, 2007. We have an investment policy
to invest these funds in liquid, investment grade securities, such as
interest-bearing money market funds, U.S. government and agency securities,
corporate notes, commercial paper, asset-backed securities and municipal
securities. Our investment portfolio does not contain securities with exposure
to sub-prime mortgages, collateralized debt obligations or auction rate
securities and we have not to date recognized an other-than-temporary impairment
on our marketable securities or any significant changes in aggregate fair value
that would impact our cash resources or liquidity. To date, we have not
experienced lack of access to our invested cash and cash equivalents; however,
we cannot provide assurances that access to our invested cash and cash
equivalents will not be impacted by adverse conditions in the financial markets.
The increase in cash, restricted cash, cash equivalents and marketable
securities in 2009 was the net result of the receipt of $45.9 million in net
proceeds in February 2009 from an underwritten public offering of our common
stock partially offset by use of cash for operations. The decrease in cash,
restricted cash, cash equivalents and marketable securities in 2008 was due to
use of cash for operations.
We estimate that our existing capital resources, interest income and
equipment financing facility will be sufficient to fund our current level of
operations through at least December 2011. However, our future capital
requirements will be substantial. Changes in our research and development plans
or other changes affecting our operating expenses or cash balances may result in
the expenditure of available resources before such time. Factors that may
require us to use our available capital resources sooner than we anticipate
include:
- continued clinical development of
our product candidates, imetelstat, GRNVAC1 and GRNOPC1;
- our ability to meaningfully reduce
manufacturing costs of current product candidates;
- future clinical trial results;
46
- progress of product and
preclinical development of our other product candidates, such as GRNCM1, GRNIC1 and
GRNCHND1;
- cost and timing of regulatory
approvals; and
- filing, maintenance, prosecution, defense and enforcement of patent claims and other intellectual property rights.
If our capital resources are insufficient to meet future capital
requirements, we will need to raise additional capital to fund our operations.
We intend to seek additional funding through strategic collaborations, public or
private equity financings, equipment loans or other financing sources that may
be available. However, we may be unable to raise sufficient additional capital
when we need it, on favorable terms or at all. If we are unable to obtain
adequate funds on reasonable terms, we may be required to curtail operations
significantly or obtain funds by entering into financing, supply or
collaboration agreements on unattractive terms or we may be required to
relinquish rights to technology or product candidates or to grant licenses on
terms that are unfavorable to us.
Cash Flows from Operating Activities
Net cash used in operations was $43.4 million, $42.0 million and $26.6
million in 2009, 2008 and 2007, respectively. The increase in net cash used for
operations in 2009 compared to 2008 was primarily the result of increased
research and development expenses associated with our clinical operations and
reduced interest income. The increase in net cash used for operations in 2008
compared to 2007 was primarily the result of reduced interest income, payments
to Biotechnology Research Corporation, our joint venture partner in TA
Therapeutics, Ltd. for scientific research services and increased clinical trial
expenses.
Cash Flows from Investing Activities
Net cash used in investing activities was $83.0 million for 2009. Net
cash provided by investing activities was $5.5 million and $16.0 million in 2008
and 2007, respectively. The decrease in cash provided by investing activities in
2009 compared to 2008 primarily reflected increased marketable securities
purchases. The decrease in net cash provided by investing activities in 2008
compared to 2007 primarily reflected reduced maturities of marketable
securities.
For the three years ended December 31, 2009, we have purchased
approximately $6.8 million in property and equipment, net of disposals, none of
which was financed through equipment financing arrangements. As of December 31,
2009, no payments were due under our equipment financing facility. As of
December 31, 2009, we had approximately $500,000 available for borrowing under
our equipment financing facility. We intend to renew the commitment for a new
equipment financing facility in 2010 to further fund equipment purchases. If we
are unable to renew the commitment, we will use our cash resources for capital
expenditures.
Cash Flows from Financing Activities
Net cash provided by financing activities in 2009 and 2007 was $51.6
million and $20.7 million, respectively. Net cash used in financing activities
in 2008 was $162,000. Net cash provided by financing activities in 2009
primarily reflected receipt of approximately $45.9 million in net proceeds from
a public offering of 7.25 million shares of our common stock at a public
offering price of $6.60 per share after deducting underwriting discounts and
commissions and offering expenses and receipt of net proceeds of $3.6 million
from the sale of 550,000 shares of Geron common stock and warrants to purchase
an additional 150,000 shares of common stock with an exercise price of $9.00 per
share to certain institutional investors. Net cash used in financing activities
in 2008 primarily reflected the repurchase of vested stock from certain
employees to provide funds for minimum payroll tax withholding requirements. Net
cash provided by financing activities in 2007 included $15.0 million in proceeds
from the exercise of warrants issued to institutional investors in connection
with a financing in December 2006 and $3.6 million in cash consideration from
the modification of certain outstanding warrants in December 2007.
47
Contractual Obligations
As of December 31, 2009 our contractual obligations for the next five
years, and thereafter were as follows:
| Principal Payments Due by Period | |||||||||||||||||||||||
| Less Than | After | ||||||||||||||||||||||
| Contractual Obligations (1) | Total | 1 Year | 1-3 Years | 4-5 Years | 5 Years | ||||||||||||||||||
| (In thousands) | |||||||||||||||||||||||
| Equipment lease | $ | 6 | $ | 6 | $ | — | $ | — | $ | — | |||||||||||||
| Operating leases (2) | — | — | — | — | — | ||||||||||||||||||
| Research funding (3) | 1,766 | 493 | 389 | 374 | 510 | ||||||||||||||||||
| Total contractual cash obligations | $ | 1,772 | $ | 499 | $ | 389 | $ | 374 | $ | 510 | |||||||||||||
| (1) | This table does not include any milestone payments under research collaborations or license agreements as the timing and likelihood of such payments are not known. In addition, this table does not include payments under our severance plan if there were a change in control of the Company or severance payments to key employees under involuntary termination. | |
| (2) | In March 2008, we issued 742,158 shares of our common stock to the lessor of our premises at 200 and 230 Constitution Drive in payment of our monthly rental obligation from August 1, 2008 through July 31, 2012. In May 2007, we issued 210,569 shares of our common stock to the lessor of our premises at 149 Commonwealth Drive in payment of our monthly rental obligation from May 1, 2007 through April 30, 2010. The fair value of the common stock issuances has been recorded as a prepaid asset and is being amortized to rent expense on a straight-line basis over the lease periods. Future minimum payments under non-cancelable operating leases are zero through July 31, 2012, as a result of the prepayments of rent with our common stock. | |
| (3) | Research funding is comprised of sponsored research commitments at various laboratories around the world. | |
Recent Accounting Pronouncements
See Note 1 of Notes to Consolidated Financial Statements for a
description of recent accounting pronouncements.
Off-Balance Sheet Arrangements
None.
ITEM 7A. QUANTITATIVE AND QUALITATIVE
DISCLOSURES ABOUT MARKET RISK
The following discussion about our market risk disclosures contains
forward-looking statements. Actual results could differ materially from those
projected in the forward-looking statements. We are exposed to market risk
related to changes in interest rates and foreign currency exchange rates. We do
not use derivative financial instruments for speculative or trading purposes.
Credit Risk. We
place our cash, restricted cash, cash equivalents and marketable securities with
six financial institutions in the United States. Deposits with banks may exceed
the amount of insurance provided on such deposits. While we monitor the cash
balances in our operating accounts and adjust the cash balances as appropriate,
these cash balances could be impacted if the underlying financial institutions
fail or could be subject to other adverse conditions in the financial markets.
Financial instruments that potentially subject us to concentrations of credit
risk consist primarily of marketable securities. Marketable securities currently
consist of U.S. Treasury securities, U.S. government-sponsored enterprise
securities and corporate notes. Our investment policy, approved by our Board of
Directors, limits the amount we may invest in any one type of investment issuer,
thereby reducing credit risk concentrations. We limit our credit and liquidity
risks through our investment policy and through regular reviews of our portfolio
against our policy. To date, we have not experienced any loss or lack of access
to cash in our operating accounts or to our cash equivalents and marketable
securities in our investment portfolios.
48
Interest Rate Risk.
The primary objective of our investment activities is to manage our marketable
securities portfolio to preserve principal and liquidity while maximizing the
return on the investment portfolio through the full investment of available
funds without significantly increasing risk. To achieve this objective,
we invest in widely diversified investments consisting
of both fixed rate and floating rate interest earning instruments, and both
carry a degree of interest rate risk. Fixed rate securities may have their fair
value adversely impacted due to a rise in interest rates, while floating rate
securities may produce less income than expected if interest rates fall. Due in
part to these factors, our future investment income may fall short of
expectations due to changes in market conditions and in interest rates or we may
suffer losses in principal if forced to sell securities which may have declined
in fair value due to changes in interest rates.
The fair value of our cash equivalents and marketable securities at
December 31, 2009 was $165.1 million. These investments include $33.4 million of
cash equivalents which are due in less than 90 days, $77.0 million of short-term
investments which are due in less than one year and $54.7 million of long-term
investments which are due in one to two years. We primarily invest our
marketable securities portfolio in short-term securities with at least an
investment grade rating to minimize interest rate and credit risk as well as to
provide for an immediate source of funds. Although changes in interest rates may
affect the fair value of the marketable securities portfolio and cause
unrealized gains or losses, such gains or losses would not be realized unless
the investments are sold. Due to the nature of our investments, which are
primarily U.S. Treasury securities, U.S. government-sponsored enterprise
securities, corporate notes and money market funds, we have concluded that there
is no material market risk exposure.
Foreign Currency Exchange Risk. Because we translate foreign currencies into
United States dollars for reporting purposes, currency fluctuations can have an
impact, though generally immaterial, on our results. We believe that our
exposure to currency exchange fluctuation risk is insignificant primarily
because our wholly-owned international subsidiary, Geron Bio-Med Ltd., satisfies
its financial obligations almost exclusively in its local currency. As of
December 31, 2009, there was an immaterial currency exchange impact from our
intercompany transactions. As of December 31, 2009, we did not engage in foreign
currency hedging activities.
ITEM 8. CONSOLIDATED FINANCIAL STATEMENTS AND
SUPPLEMENTARY DATA
The following consolidated financial statements and the related notes
thereto, of Geron Corporation and the Report of Independent Registered Public
Accounting Firm, Ernst & Young LLP, are filed as a part of this Form
10-K.
| Page | ||
| Report of Independent Registered Public Accounting Firm | 50 | |
| Consolidated Balance Sheets | 51 | |
| Consolidated Statements of Operations | 52 | |
| Consolidated Statements of Stockholders’ Equity | 53 | |
| Consolidated Statements of Cash Flows | 54 | |
| Notes to Consolidated Financial Statements | 55 | |
| Supplemental Data: Quarterly Financial Information | 76 |
49
REPORT OF INDEPENDENT REGISTERED PUBLIC
ACCOUNTING FIRM
The Board of Directors
and Stockholders of Geron Corporation
We have audited the
accompanying consolidated balance sheets of Geron Corporation as of December 31,
2009 and 2008, and the related consolidated statements of operations,
stockholders’ equity, and cash flows for each of the three years in the period
ended December 31, 2009. These financial statements are the responsibility of
the Company’s management. Our responsibility is to express an opinion on these
financial statements based on our audits.
We conducted our audits
in accordance with the standards of the Public Company Accounting Oversight
Board (United States). Those standards require that we plan and perform the
audit to obtain reasonable assurance about whether the financial statements are
free of material misstatement. An audit includes examining, on a test basis,
evidence supporting the amounts and disclosures in the financial statements. An
audit also includes assessing the accounting principles used and significant
estimates made by management, as well as evaluating the overall financial
statement presentation. We believe that our audits provide a reasonable basis
for our opinion.
In our opinion, the
financial statements referred to above present fairly, in all material respects,
the consolidated financial position of Geron Corporation at December 31, 2009
and 2008, and the consolidated results of its operations and its cash flows for
each of the three years in the period ended December 31, 2009, in conformity
with U.S. generally accepted accounting principles.
We also have audited, in
accordance with the standards of the Public Company Accounting Oversight Board
(United States), Geron Corporation’s internal control over financial reporting
as of December 31, 2009, based on criteria established in Internal
Control-Integrated Framework issued by the Committee of Sponsoring Organizations
of the Treadway Commission and our report dated February 26, 2010 expressed an
unqualified opinion thereon.
/s/ Ernst & Young
LLP
Palo Alto, California
February 26, 2010
February 26, 2010
50
GERON CORPORATION
CONSOLIDATED BALANCE
SHEETS
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| (In thousands, except share | ||||||||
| and per share data) | ||||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 34,601 | $ | 109,348 | ||||
| Restricted cash | 791 | 816 | ||||||
| Current portion of marketable securities | 77,009 | 53,491 | ||||||
| Interest and other receivables | 1,318 | 882 | ||||||
| Current portion of prepaid assets | 4,060 | 3,709 | ||||||
| Total current assets | 117,779 | 168,246 | ||||||
| Noncurrent portion of marketable securities | 54,669 | — | ||||||
| Noncurrent portion of prepaid assets | 2,372 | 2,236 | ||||||
| Investments in licensees | 1,328 | 657 | ||||||
| Property and equipment, net | 3,938 | 4,386 | ||||||
| Deposits and other assets | 296 | 693 | ||||||
| $ | 180,382 | $ | 176,218 | |||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 2,176 | $ | 2,414 | ||||
| Accrued compensation | 1,757 | 1,398 | ||||||
| Accrued liabilities (including amounts for related parties: | ||||||||
| 2009-none, 2008-$270) | 1,925 | 2,248 | ||||||
| Current portion of deferred revenue | 700 | 27 | ||||||
| Current portion of advance payment from related party for research and | ||||||||
| development, net | — | 440 | ||||||
| Fair value of derivatives | 897 | 1,184 | ||||||
| Total current liabilities | 7,455 | 7,711 | ||||||
| Noncurrent portion of deferred revenue | 350 | 52 | ||||||
| Commitments and contingencies | ||||||||
| Stockholders’ equity: | ||||||||
| Preferred stock, $0.001 par value; 3,000,000 shares authorized; no shares | ||||||||
| issued and outstanding at December 31, 2009 and 2008 | — | — | ||||||
| Common stock, $0.001 par value; 200,000,000 shares authorized; | ||||||||
| 92,521,946 and 81,070,464 shares issued and outstanding at | ||||||||
| December 31, 2009 and 2008, respectively | 92 | 81 | ||||||
| Additional paid-in capital | 750,158 | 675,227 | ||||||
| Accumulated deficit | (577,267 | ) | (506,893 | ) | ||||
| Accumulated other comprehensive (loss) income | (406 | ) | 40 | |||||
| Total stockholders’ equity | 172,577 | 168,455 | ||||||
| $ | 180,382 | $ | 176,218 | |||||
See accompanying
notes.
51
GERON CORPORATION
CONSOLIDATED STATEMENTS OF
OPERATIONS
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (In thousands, except share and per share data) | ||||||||||||
| Revenues from collaborative agreements (including | ||||||||||||
| amounts from related parties: 2009-none, 2008-$79, | ||||||||||||
| 2007-$487) | $ | 450 | $ | 294 | $ | 672 | ||||||
| License fees and royalties (including amounts from related | ||||||||||||
| parties: 2009-none, 2008-$1,500, 2007-none) | 1,276 | 2,509 | 6,950 | |||||||||
| Total revenues | 1,726 | 2,803 | 7,622 | |||||||||
| Operating expenses: | ||||||||||||
| Research and development (including amounts | ||||||||||||
| for related parties: 2009-$1,755, 2008-$794, | ||||||||||||
| 2007-$941) | 57,617 | 53,664 | 54,624 | |||||||||
| General and administrative | 14,343 | 16,183 | 15,837 | |||||||||
| Total operating expenses | 71,960 | 69,847 | 70,461 | |||||||||
| Loss from operations | (70,234 | ) | (67,044 | ) | (62,839 | ) | ||||||
| Unrealized gain on fair value of derivatives | 157 | 418 | 15,453 | |||||||||
| Interest and other income | 1,374 | 5,542 | 10,791 | |||||||||
| Losses recognized under equity method investment | (1,338 | ) | (844 | ) | — | |||||||
| Interest and other expense | (143 | ) | (93 | ) | (102 | ) | ||||||
| Net loss | (70,184 | ) | (62,021 | ) | (36,697 | ) | ||||||
| Deemed dividend on derivatives | (190 | ) | — | (9,081 | ) | |||||||
| Net loss applicable to common stockholders | $ | (70,374 | ) | $ | (62,021 | ) | $ | (45,778 | ) | |||
| Basic and diluted net loss per share applicable to common | ||||||||||||
| stockholders: | ||||||||||||
| Net loss per share applicable to common stockholders | $ | (0.80 | ) | $ | (0.79 | ) | $ | (0.62 | ) | |||
| Shares used in computing net loss per share applicable to | ||||||||||||
| common stockholders | 88,078,557 | 78,187,795 | 74,206,249 | |||||||||
See accompanying
notes.
52
GERON CORPORATION
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’
EQUITY
| Accumu- | ||||||||||||||||||||
| Additional | Accumu- | lated Other | Total | |||||||||||||||||
| Common Stock | Paid-In | lated | Comprehensive | Stockholders’ | ||||||||||||||||
| Shares | Amount | Capital | Deficit | Income (Loss) | Equity | |||||||||||||||
| (In thousands, except share data) | ||||||||||||||||||||
| Balances at December 31, 2006 | 70,449,058 | $ | 70 | $ | 573,156 | $ | (399,094 | ) | $ | (213 | ) | $ | 173,919 | |||||||
| Net loss | — | — | — | (36,697 | ) | — | (36,697 | ) | ||||||||||||
| Net change in unrealized gain (loss) on marketable | ||||||||||||||||||||
| securities and investments in licensees | — | — | — | — | 235 | 235 | ||||||||||||||
| Cumulative translation adjustment | — | — | — | — | 11 | 11 | ||||||||||||||
| Comprehensive loss | (36,451 | ) | ||||||||||||||||||
| Reclassification of fair value of derivatives, net | — | — | 21,974 | — | — | 21,974 | ||||||||||||||
| Deemed dividend in connection with warrants to purchase | ||||||||||||||||||||
| common stock, including cash consideration | — | — | 12,711 | (9,081 | ) | — | 3,630 | |||||||||||||
| Stock-based compensation related to issuance of | ||||||||||||||||||||
| common stock and options in exchange for | ||||||||||||||||||||
| services | 1,169,823 | 1 | 10,149 | — | — | 10,150 | ||||||||||||||
| Issuance of common stock upon exercise of warrants | 3,470,204 | 4 | 15,147 | — | — | 15,151 | ||||||||||||||
| Issuance of common stock under employee stock | ||||||||||||||||||||
| plans, net | 881,985 | 1 | 4,870 | — | — | 4,871 | ||||||||||||||
| Stock-based compensation for equity-based awards | ||||||||||||||||||||
| to employees and directors | — | — | 11,367 | — | — | 11,367 | ||||||||||||||
| 401(k) contribution | 91,369 | — | 1,063 | — | — | 1,063 | ||||||||||||||
| Balances at December 31, 2007 | 76,062,439 | 76 | 650,437 | (444,872 | ) | 33 | 205,674 | |||||||||||||
| Net loss | — | — | — | (62,021 | ) | — | (62,021 | ) | ||||||||||||
| Net change in unrealized gain (loss) on marketable | ||||||||||||||||||||
| securities and investments in licensees | — | — | — | — | 16 | 16 | ||||||||||||||
| Cumulative translation adjustment | — | — | — | — | (9 | ) | (9 | ) | ||||||||||||
| Comprehensive loss | (62,014 | ) | ||||||||||||||||||
| Stock-based compensation related to issuance | ||||||||||||||||||||
| of common stock in exchange for services | 2,294,685 | 2 | 9,789 | — | — | 9,791 | ||||||||||||||
| Issuance of common stock under employee stock | ||||||||||||||||||||
| plans, net | 2,506,424 | 3 | 2,463 | — | — | 2,466 | ||||||||||||||
| Stock-based compensation for equity-based awards | ||||||||||||||||||||
| to employees and directors | — | — | 11,493 | — | — | 11,493 | ||||||||||||||
| 401(k) contribution | 206,916 | — | 1,045 | — | — | 1,045 | ||||||||||||||
| Balances at December 31, 2008 | 81,070,464 | 81 | 675,227 | (506,893 | ) | 40 | 168,455 | |||||||||||||
| Net loss | — | — | — | (70,184 | ) | — | (70,184 | ) | ||||||||||||
| Net change in unrealized gain (loss) on marketable | ||||||||||||||||||||
| securities and investments in licensees | — | — | — | — | (445 | ) | (445 | ) | ||||||||||||
| Cumulative translation adjustment | — | — | — | — | (1 | ) | (1 | ) | ||||||||||||
| Comprehensive loss | (70,630 | ) | ||||||||||||||||||
| Issuance of common stock in connection with public | ||||||||||||||||||||
| offering, net of issuance costs of $1,916 | 7,250,000 | 7 | 45,926 | — | — | 45,933 | ||||||||||||||
| Issuance of common stock in connection with private | ||||||||||||||||||||
| offering, net of issuance costs of $18 | 550,000 | 1 | 3,584 | — | — | 3,585 | ||||||||||||||
| Reclassification of fair value of derivatives, net | — | — | 130 | — | — | 130 | ||||||||||||||
| Deemed dividend in connection with amendments to warrants | ||||||||||||||||||||
| to purchase common stock | — | — | 190 | (190 | ) | — | — | |||||||||||||
| Stock-based compensation related to issuance | ||||||||||||||||||||
| of common stock and options in exchange for services | 1,272,438 | 1 | 8,114 | — | — | 8,115 | ||||||||||||||
| Issuance of common stock under employee stock | ||||||||||||||||||||
| plans, net | 2,110,418 | 2 | 5,253 | — | — | 5,255 | ||||||||||||||
| Stock-based compensation for equity-based awards | ||||||||||||||||||||
| to employees and directors | — | — | 10,575 | — | — | 10,575 | ||||||||||||||
| 401(k) contribution | 268,626 | — | 1,159 | — | — | 1,159 | ||||||||||||||
| Balances at December 31, 2009 | 92,521,946 | $ | 92 | $ | 750,158 | $ | (577,267 | ) | $ | (406 | ) | $ | 172,577 | |||||||
See accompanying
notes.
53
GERON CORPORATION
CONSOLIDATED STATEMENTS OF CASH
FLOWS
| Year Ended December 31, | ||||||||||||
| 2009 | 2008 | 2007 | ||||||||||
| (In thousands) | ||||||||||||
| Cash flows from operating activities | ||||||||||||
| Net loss | $ | (70,184 | ) | $ | (62,021 | ) | $ | (36,697 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
| Depreciation and amortization | 1,753 | 2,017 | 1,667 | |||||||||
| Accretion and amortization on investments, net | 926 | (1,153 | ) | (3,227 | ) | |||||||
| Loss (gain) on retirement/sale of property and equipment | 130 | (6 | ) | — | ||||||||
| Issuance of common stock and warrants in exchange for services | ||||||||||||
| by non-employees | 4,866 | 2,068 | 5,674 | |||||||||
| Stock-based compensation for employees and directors | 10,575 | 11,493 | 11,367 | |||||||||
| Amortization related to 401(k) contributions | 494 | 405 | 263 | |||||||||
| Loss on investments in licensees | 1,364 | 887 | 106 | |||||||||
| Unrealized gain on fair value of derivatives | (157 | ) | (418 | ) | (15,453 | ) | ||||||
| Changes in assets and liabilities: | ||||||||||||
| Interest and other receivables | (436 | ) | (94 | ) | 487 | |||||||
| Prepaid assets | 3,019 | 6,394 | 2,583 | |||||||||
| Investments in licensees | — | — | 5 | |||||||||
| Deposits and other assets | (99 | ) | 2 | (371 | ) | |||||||
| Accounts payable | (56 | ) | (443 | ) | 898 | |||||||
| Accrued compensation | 4,166 | 2,332 | 2,855 | |||||||||
| Accrued liabilities | (265 | ) | (1,912 | ) | 2,418 | |||||||
| Deferred revenue | 971 | (240 | ) | (945 | ) | |||||||
| Advance payment from related party for research and development | (440 | ) | (1,287 | ) | 1,727 | |||||||
| Translation adjustment | (1 | ) | (9 | ) | 11 | |||||||
| Net cash used in operating activities | (43,374 | ) | (41,985 | ) | (26,632 | ) | ||||||
| Cash flows from investing activities | ||||||||||||
| Restricted cash transfer | 25 | 1,624 | (1,910 | ) | ||||||||
| Loan to related party | — | (1,500 | ) | — | ||||||||
| Investment in licensee, net | (2,009 | ) | — | — | ||||||||
| Proceeds from sale of property and equipment | — | 15 | — | |||||||||
| Purchases of property and equipment | (1,435 | ) | (2,337 | ) | (2,990 | ) | ||||||
| Purchases of marketable securities | (200,109 | ) | (78,332 | ) | (154,876 | ) | ||||||
| Proceeds from maturities of marketable securities | 120,524 | 86,000 | 175,816 | |||||||||
| Proceeds from sale of investment in licensees | 1 | — | — | |||||||||
| Net cash (used in) provided by investing activities | (83,003 | ) | 5,470 | 16,040 | ||||||||
| Cash flows from financing activities | ||||||||||||
| Repurchase of common stock | — | (455 | ) | — | ||||||||
| Proceeds from issuance of common stock and warrants, net of issuance costs | 51,630 | 293 | 20,735 | |||||||||
| Net cash provided by (used in) financing activities | 51,630 | (162 | ) | 20,735 | ||||||||
| Net (decrease) increase in cash and cash equivalents | (74,747 | ) | (36,677 | ) | 10,143 | |||||||
| Cash and cash equivalents, at beginning of year | 109,348 | 146,025 | 135,882 | |||||||||
| Cash and cash equivalents, at end of year | $ | 34,601 | $ | 109,348 | $ | 146,025 | ||||||
See accompanying
notes.
54
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. ORGANIZATION AND SUMMARY OF SIGNIFICANT
ACCOUNTING POLICIES
Organization
Geron Corporation (“we” or “Geron”) was incorporated in the State of
Delaware on November 29, 1990. We are a biopharmaceutical company that is
developing first-in-class biopharmaceuticals for the treatment of cancer and
chronic degenerative diseases, including spinal cord injury, heart failure and
diabetes. We are advancing an anti-cancer drug and a cancer vaccine that target
the enzyme telomerase through multiple clinical trials in different cancers. The
products are based on our core expertise in telomerase and human embryonic stem
cells. Principal activities to date have included obtaining financing, securing
operating facilities and conducting research and development. We have no
therapeutic products currently available for sale and do not expect to have any
therapeutic products commercially available for sale for a period of years, if
at all. These factors indicate that our ability to continue research and
development activities is dependent upon the ability of our management to obtain
additional financing as required.
Principles of
Consolidation
The consolidated financial statements include the accounts of Geron, our
wholly-owned subsidiary, Geron Bio-Med Ltd. (Geron Bio-Med), a United Kingdom
company, and our majority-owned subsidiary, TA Therapeutics, Ltd. (TAT), a Hong
Kong company. We have eliminated intercompany accounts and transactions. We
prepare the financial statements of Geron Bio-Med using the local currency as
the functional currency. We translate the assets and liabilities of Geron
Bio-Med at rates of exchange at the balance sheet date and translate income and
expense items at average monthly rates of exchange. The resultant translation
adjustments are included in accumulated other comprehensive income (loss), a
separate component of stockholders’ equity. The functional currency for TAT is
U.S. dollars.
Net Loss Per Share
Basic earnings (loss) per share is calculated based on the weighted
average number of shares of common stock outstanding during the period. Diluted
earnings (loss) per share is calculated based on the weighted average number of
shares of common stock and dilutive securities outstanding during the period.
Potential dilutive securities primarily consist of outstanding employee stock
options, restricted stock and warrants to purchase common stock and have been
determined using the treasury stock method at an average market price during the
period.
Because we were in a net loss position, diluted earnings per share
excludes the effects of potential dilutive securities. Had we been in a net
income position, diluted earnings per share would have included the shares used
in the computation of basic net loss per share as well as an additional
1,260,417, 300,011 and 2,063,459 shares for 2009, 2008 and 2007, respectively,
related to outstanding options, restricted stock and warrants (as determined
using the treasury stock method at the estimated average market
value).
Use of Estimates
The preparation of financial statements in conformity with accounting
principles generally accepted in the United States requires us to make estimates
and assumptions that affect the amounts reported in the financial statements and
accompanying notes. On a regular basis, management evaluates these estimates and
assumptions. Actual results could differ from those estimates.
Fair Value of Financial
Instruments
Cash Equivalents and Marketable Securities
We consider all highly liquid investments with an original maturity of
three months or less to be cash equivalents. We are subject to credit risk
related to our cash equivalents and marketable securities. We place our cash and
cash equivalents in money market funds. Our investments include U.S. Treasury
securities, U.S. government-sponsored enterprise securities and corporate notes
with original maturities ranging from four to 24 months.
55
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
We classify our marketable debt securities as available-for-sale. We
record available-for-sale securities at fair value with unrealized gains and
losses reported in accumulated other comprehensive income (loss) in
stockholders’ equity. Realized gains and losses are included in interest and
other income and are derived using the specific identification method for
determining the cost of securities sold and have been insignificant to date.
Dividend and interest income are recognized when earned and included in interest
and other income in our consolidated statements of operations. We recognize a
charge when the declines in the fair values of our available-for-sale securities
below the amortized cost basis are judged to be other-than-temporary. We
consider various factors in determining whether to recognize an
other-than-temporary charge, including whether we intend to sell the security or
whether it is more likely than not that we would be required to sell the
security. Declines in market value associated with credit losses judged as
other-than-temporary result in a charge to interest and other income.
Other-than-temporary charges not related to credit losses are included in
accumulated other comprehensive income (loss) in stockholders’ equity. No
other-than-temporary impairment charges were recorded for our available-for-sale
securities for the years ended December 31, 2009, 2008 and 2007. See Note 2 on
Fair Value Measurements.
Marketable and Non-Marketable Investments in Licensees
Investments in non-marketable nonpublic companies, in which we own less
than 20% of the outstanding voting stock and do not otherwise have the ability
to exert significant influence over the investees, are carried at cost, as
adjusted for other-than-temporary impairments. Investments in marketable equity
securities are carried at fair value as of the balance sheet date with
unrealized gains and losses reported in accumulated other comprehensive income
(loss) in stockholders’ equity. Realized gains or losses are included in
interest and other income and are derived using the specific identification
method.
We apply the equity method of accounting for investments in licensees in
which we own more than 20% of the outstanding voting stock or otherwise have the
ability to exert significant influence over the investees. Under this method, we
increase (decrease) the carrying value of our investment by a proportionate
share of the investee’s earnings (losses). If losses exceed the carrying value
of the investment, losses are then applied against any advances to the investee,
including any commitment to provide financial support, until those amounts are
reduced to zero. The equity method is then suspended until the investee has
earnings. Any proportionate share of investee earnings is first applied to the
share of accumulated losses not recognized during the period the equity method
was suspended.
We monitor our investments in licensees for impairment on a quarterly
basis and make appropriate reductions in carrying values when such impairments
are determined to be other-than-temporary. Other-than-temporary charges are
included in interest and other income. Factors used in determining whether an
other-than-temporary charge should be recognized include, but are not limited
to, the current business environment including competition and uncertainty of
financial condition; going concern considerations such as the rate at which the
investee company utilizes cash, and the investee company’s ability to obtain
additional private financing to fulfill its stated business plan; the need for
changes to the investee company’s existing business model due to changing
business environments and its ability to successfully implement necessary
changes; and the general progress toward product development, including clinical
trial results. See Note 2 on Fair Value Measurements.
Fair Value of Derivatives
For warrants and non-employee options classified as assets or
liabilities, the fair value of these instruments is recorded on the consolidated
balance sheet at inception of such classification and adjusted to fair value at
each financial reporting date. The change in fair value of the warrants and
non-employee options is recorded in the consolidated statements of operations as
unrealized gain (loss) on fair value of derivatives. Fair value of warrants and
non-employee options is estimated using the Black Scholes option-pricing model.
The warrants and non-employee options continue to be reported as an asset or
liability until such time as the instruments are exercised or expire or are
otherwise modified to remove the provisions which require this treatment, at
which time these instruments are marked to fair value and reclassified from
assets or liabilities to stockholders’ equity. For warrants and non-employee
options classified as permanent equity, the fair value of the warrants and
non-employee options is recorded in stockholders’ equity and no further
adjustments are made. See Note 2 on Fair Value Measurements.
56
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Revenue Recognition
We have several license agreements with various oncology, diagnostics,
research tools, agriculture and biologics production companies. With certain of
these agreements, we receive nonrefundable license payments in cash or equity
securities, option payments in cash or equity securities, royalties on future
sales of products, milestone payments, or any combination of these items.
Upfront nonrefundable signing, license or non-exclusive option fees are
recognized as revenue when rights to use the intellectual property related to
the license have been delivered and over the term of the agreement if we have
continuing performance obligations. Milestone payments, which are subject to
substantive contingencies, are recognized upon completion of specified
milestones, representing the culmination of the earnings process, according to
contract terms. Royalties are generally recognized upon receipt of the related
royalty payment. Deferred revenue represents the portion of research and license
payments received which has not been earned. When payments are received in
equity securities, we do not recognize any revenue unless such securities are
determined to be realizable in cash.
We recognize revenue under collaborative agreements as the related
research and development costs for services are rendered. We recognize related
party revenue under collaborative agreements as the related research and
development costs for services are rendered and when the source of funds have
not been derived from our contributions to the related party.
Restricted Cash
The components of restricted cash
are as follows:
| December 31, | ||||||
| 2009 | 2008 | |||||
| (In thousands) | ||||||
| Certificate of deposit for unused equipment line of credit | $ | 530 | $ | 530 | ||
| Certificate of deposit for credit card purchases | 261 | 258 | ||||
| Funds held in trust for creditors of TA Therapeutics, Ltd. | — | 28 | ||||
| $ | 791 | $ | 816 | |||
Research and Development Expenses
All research and development costs are expensed as incurred. The value of
acquired in-process research and development is charged to research and
development expense on the date of acquisition, if not acquired in connection
with a business combination. Research and development expenses include, but are
not limited to, acquired in-process technology deemed to have no alternative
future use, payroll and personnel expense, lab supplies, preclinical studies,
raw materials to manufacture clinical trial drugs, manufacturing costs for
research and clinical trial materials, sponsored research at other labs,
consulting, costs to maintain technology licenses and research-related
overhead.
Depreciation and Amortization
We record property and equipment at cost and calculate depreciation using
the straight-line method over the estimated useful lives of the assets,
generally four years. Leasehold improvements are amortized over the shorter of
the estimated useful life or remaining term of the lease.
Stock-Based Compensation
Geron maintains various stock incentive plans under which stock options
and restricted stock awards are granted to employees, non-employee members of
the Board of Directors and consultants. We also have an employee stock purchase
plan for all eligible employees. We recognize compensation expense on a
straight-line basis for stock-based awards granted after January 1, 2006, plus
unvested awards granted prior to January 1, 2006 based on the grant-date fair
value estimated using accounting guidance in effect at that time and following
the straight-line attribution method.
57
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
We use the Black Scholes option-pricing valuation model to estimate the
grant-date fair value of our stock-based awards. For additional information, see
Note 8 on Stockholders’ Equity. The determination of fair value for stock-based
awards on the date of grant using an option-pricing model is affected by our
stock price as well as assumptions regarding a number of complex and subjective
variables. These variables include, but are not limited to, our expected stock
price volatility over the term of the awards and actual and projected employee
exercise behaviors. The stock-based compensation expense related to restricted
stock awards is determined using the fair value of Geron common stock on the
date of grant and reduced for estimated forfeitures as applicable. The fair
value is amortized as compensation expense over the service period of the award
on a straight-line basis.
For our non-employee stock-based awards, the measurement date on which
the fair value of the stock-based award is calculated is equal to the earlier of
(i) the date at which a commitment for performance by the counterparty to earn
the equity instrument is reached or (ii) the date at which the counterparty’s
performance is complete. We recognize stock-based compensation expense for the
fair value of the vested portion of non-employee awards in our consolidated
statements of operations.
Comprehensive Loss
Comprehensive loss is comprised of net loss and other comprehensive
income (loss). Other comprehensive income (loss) includes certain changes in
stockholders’ equity which are excluded from net loss.
The components of accumulated other
comprehensive income (loss) are as follows:
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| (In thousands) | ||||||||
| Unrealized (loss) gain on available-for-sale securities and | ||||||||
| marketable investments in licensees | $ | (234 | ) | $ | 211 | |||
| Foreign currency translation adjustments | (172 | ) | (171 | ) | ||||
| $ | (406 | ) | $ | 40 | ||||
In 2009 and 2008, we recognized other-than-temporary impairment charges
of none and $43,000, respectively, related to our investments in licensees. In
addition, $26,000 and none of previously unrecognized unrealized loss was
eliminated from accumulated other comprehensive income (loss) in 2009 and 2008,
respectively. See Note 2 on Fair Value Measurements.
Income Taxes
We maintain deferred tax assets and liabilities that reflect the net tax
effects of temporary differences between the carrying amounts of assets and
liabilities for financial reporting purposes and the amounts used for income tax
purposes and are subject to tests of recoverability. Our deferred tax assets
include net operating loss carryforwards, research credits and capitalized
research and development. Deferred tax assets and liabilities are measured using
enacted tax rates expected to apply to taxable income in the years in which
those temporary differences are expected to be recovered or settled. Our net
deferred tax asset has been fully offset by a valuation allowance because of our
history of losses. Any potential accrued interest and penalties related to
unrecognized tax benefits within operations would be recorded as income tax
expense. To date, there have been no interest or penalties charged to us related
to the underpayment of income taxes.
Concentrations of Customers and Suppliers
The majority of our revenues was earned in the United States. One new
customer accounted for 46% of our 2009 revenues. One related party customer
accounted for approximately 54% of our 2008 revenues. One existing customer
accounted for 79% of our 2007 revenues.
We contract third-party manufacturers to produce GMP-grade drugs and
vaccines for preclinical and clinical studies. We also contract for raw
materials to supply those manufacturers. Certain development and clinical
activities may be delayed if we are unable to obtain sufficient quantities of
raw materials or GMP-grade drugs and vaccines from our third-party sources or
other third-party sources.
58
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Recent Accounting
Pronouncement
In October 2009, the Financial Accounting Standards Board issued
authoritative guidance addressing revenue arrangements with multiple
deliverables. The guidance eliminates the criterion for objective and reliable
evidence of fair value for the undelivered products or services. Instead,
revenue arrangements with multiple deliverables should be divided into separate
units provided the deliverables meet certain criteria. This guidance also
eliminates the use of the residual method of allocation and requires that the
arrangement consideration be allocated at the inception of the arrangement to
all deliverables based on their relative selling price. The guidance also
provides a hierarchy for estimating the selling price of each of the
deliverables. The new guidance is applicable prospectively for any arrangements
we enter into after January 1, 2011. We are evaluating the potential impact, if
any, of this new guidance on our consolidated financial statements.
2. FAIR VALUE MEASUREMENTS
We categorize assets and liabilities recorded at fair value on our
consolidated balance sheet based upon the level of judgment associated with
inputs used to measure their fair value. The categories are as
follows:
|
Level
1
|
Inputs are
unadjusted, quoted prices in active markets for identical assets or
liabilities at the measurement date. An active market for the asset or
liability is a market in which transactions for the asset or liability
occur with sufficient frequency and volume to provide pricing information
on an ongoing basis.
|
||
|
Level
2
|
Inputs (other than
quoted market prices included in Level 1) are either directly or
indirectly observable for the asset or liability through correlation with
market data at the measurement date and for the duration of the
instrument’s anticipated life.
|
||
|
Level
3
|
Inputs reflect
management’s best estimate of what market participants would use in
pricing the asset or liability at the measurement date. Consideration is
given to the risk inherent in the valuation technique and the risk
inherent in the inputs to the
model.
|
A financial instrument’s categorization within the valuation hierarchy is
based upon the lowest level of input that is significant to the fair value
measurement. Following is a description of the valuation methodologies used for
instruments measured at fair value on our consolidated balance sheet, including
the category for such instruments.
Cash Equivalents and Marketable Securities
Available-for-Sale
Where quoted prices are available in an active market, securities are
categorized as Level 1. Examples of such Level 1 securities include highly
liquid U.S. Treasury securities, U.S. government-sponsored enterprise securities
and money market funds. If quoted market prices are not available for the
specific security, then fair values are estimated by using pricing models,
quoted prices of securities with similar characteristics or discounted cash
flows. Examples of such Level 2 instruments include corporate notes,
asset-backed securities and commercial paper.
59
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Marketable securities by security
type at December 31, 2009 were as follows:
| Gross | Gross | ||||||||||||
| Unrealized | Unrealized | Estimated | |||||||||||
| Cost | Gains | Losses | Fair Value | ||||||||||
| (In thousands) | |||||||||||||
| Included in cash and cash equivalents: | |||||||||||||
| Money market funds | $ | 33,395 | $ | — | $ | — | $ | 33,395 | |||||
| Restricted cash: | |||||||||||||
| Certificates of deposit | $ | 791 | $ | — | $ | — | $ | 791 | |||||
| Marketable securities: | |||||||||||||
| U.S. Treasury securities (due in less than 1 year) | $ | 58,146 | $ | 20 | $ | (7 | ) | $ | 58,159 | ||||
| Government-sponsored enterprise securities (due in | |||||||||||||
| 1 to 2 years) | 14,058 | — | (37 | ) | 14,021 | ||||||||
| Corporate notes (due in less than 1 year) | 18,847 | 11 | (8 | ) | 18,850 | ||||||||
| Corporate notes (due in 1 to 2 years) | 40,861 | — | (213 | ) | 40,648 | ||||||||
| $ | 131,912 | $ | 31 | $ | (265 | ) | $ | 131,678 | |||||
Marketable securities by security
type at December 31, 2008 were as follows:
| Gross | Gross | ||||||||||||
| Unrealized | Unrealized | Estimated | |||||||||||
| Cost | Gains | Losses | Fair Value | ||||||||||
| (In thousands) | |||||||||||||
| Included in cash and cash equivalents: | |||||||||||||
| Money market funds | $ | 106,046 | $ | — | $ | — | $ | 106,046 | |||||
| U.S. Treasury securities | 1,254 | — | — | 1,254 | |||||||||
| $ | 107,300 | $ | — | $ | — | $ | 107,300 | ||||||
| Restricted cash: | |||||||||||||
| Certificates of deposit | $ | 788 | $ | — | $ | — | $ | 788 | |||||
| Money market funds | 28 | — | — | 28 | |||||||||
| $ | 816 | $ | — | $ | — | $ | 816 | ||||||
| Marketable securities: | |||||||||||||
| U.S. Treasury securities (due in less than 1 year) | $ | 10,314 | $ | 55 | $ | — | $ | 10,369 | |||||
| Government-sponsored enterprise securities (due in | |||||||||||||
| less than 1 year) | 25,764 | 87 | — | 25,851 | |||||||||
| Commercial paper (due in less than 1 year) | 17,176 | 95 | — | 17,271 | |||||||||
| Investments in licensees | 27 | — | (26 | ) | 1 | ||||||||
| $ | 53,281 | $ | 237 | $ | (26 | ) | $ | 53,492 | |||||
Marketable securities with
unrealized losses at December 31, 2009 and 2008 were as follows:
| Less Than 12 Months | 12 Months or Greater | Total | |||||||||||||||||||
| Gross | Gross | Gross | |||||||||||||||||||
| Estimated | Unrealized | Estimated | Unrealized | Estimated | Unrealized | ||||||||||||||||
| Fair Value | Losses | Fair Value | Losses | Fair Value | Losses | ||||||||||||||||
| (In thousands) | |||||||||||||||||||||
| As of December 31, 2009: | |||||||||||||||||||||
| U.S. Treasury securities (due in less | |||||||||||||||||||||
| than 1 year) | $ | 18,859 | $ | (7 | ) | $ | — | $ | — | $ | 18,859 | $ | (7 | ) | |||||||
| Government-sponsored enterprise | |||||||||||||||||||||
| securities (due in 1 to 2 years) | 14,021 | (37 | ) | — | — | 14,021 | (37 | ) | |||||||||||||
| Corporate notes (due in less than 1 year) | 7,524 | (8 | ) | — | — | 7,524 | (8 | ) | |||||||||||||
| Corporate notes (due in 1 to 2 years) | 40,648 | (213 | ) | — | — | 40,648 | (213 | ) | |||||||||||||
| $ | 81,052 | $ | (265 | ) | $ | — | $ | — | $ | 81,052 | $ | (265 | ) | ||||||||
| As of December 31, 2008: | |||||||||||||||||||||
| Investments in licensees | $ | — | $ | — | $ | 1 | $ | (26 | ) | $ | 1 | $ | (26 | ) | |||||||
60
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The gross unrealized losses related to U.S Treasury securities,
government-sponsored enterprise securities and corporate notes as of December
31, 2009 were due to changes in interest rates. The gross unrealized losses
related to investments in licensees as of December 31, 2008 were a result of
declining valuations for those biopharmaceutical companies. We determined that
the gross unrealized losses on our marketable securities as of December 31, 2009
and 2008 were temporary in nature. We review our investments quarterly to
identify and evaluate whether any investments have indications of possible
impairment. Factors considered in determining whether a loss is temporary
include the length of time and extent to which the fair value has been less than
the cost basis, the financial condition and near-term prospects of the investee,
and whether we intend to sell the security or whether it is more likely than not
that we would be required to sell the security.
Marketable and Non-Marketable Investments in
Licensees
Where quoted prices are available in an active market, securities are
categorized as Level 1. Level 1 securities include publicly traded equities.
Significant investments in licensees accounted for using the equity method of
accounting or equity securities in non-marketable companies are not measured at
fair value and are not assigned a category level.
We recognized charges of none, $43,000 and $106,000 in 2009, 2008 and
2007, respectively, related to other-than-temporary declines in the fair values
of certain of our investments in licensees. As of December 31, 2009 and 2008,
the carrying values of our investments in non-marketable nonpublic companies
were $1,328,000 and $656,000, respectively. We recognized net realized losses of
$26,000 for 2009 and net realized gains of $1,000 for 2007 related to sales of
investments in licensees. In connection with the sales, $26,000 of previously
unrecognized unrealized loss and $2,000 of previously unrecognized unrealized
gain was eliminated from accumulated other comprehensive income (loss) for 2009
and 2007, respectively. No sales of investments in licensees occurred in 2008.
See Note 3 on Joint Venture and Related Party Transactions for further
discussion of investments in licensees.
Derivatives
Warrants to purchase common stock and non-employee options are normally
traded less actively, have trade activity that is one way, and/or traded in
less-developed markets and are therefore valued based upon models with
significant unobservable market parameters, resulting in Level 3
categorization.
The fair value of derivatives has been calculated at each reporting date
using the Black Scholes option-pricing model with the following
assumptions:
| December 31, | ||||
| 2009 | 2008 | |||
| Dividend yield | None | None | ||
| Expected volatility range | 0.607 to 0.632 | 0.749 to 0.758 | ||
| Risk-free interest rate range | 0.06% to 2.69% | 0.57% to 1.71% | ||
| Expected term | 4 mos to 5 yrs | 1 yr to 6 yrs | ||
The expected volatility range is based on historical volatilities of our
stock since traded options on Geron stock do not correspond to derivatives’
terms and trading volume of Geron options is limited. The expected term of
derivatives is equal to the remaining contractual term of the instrument. The
risk-free interest rate range is based on the U.S. Zero Coupon Treasury Strip
Yields for the expected term in effect on the reporting date. Dividend yield is
based on historical cash dividend payments, which have been none to date.
As of December 31, 2009 and 2008, the following warrants and non-employee
options to purchase common stock were considered derivatives and classified as
current liabilities:
| Number of Shares at | Fair Value at | ||||||||||||||||
| Exercise | December 31, | December 31, | |||||||||||||||
| Issuance Date | Price | 2009 | 2008 | Exercisable Date | Expiration Date | 2009 | 2008 | ||||||||||
| (In thousands) | |||||||||||||||||
| April 2005 | $ | 7.95 | 351,852 | 351,852 | April 2005 | April 2010 | $ | 58 | $ | 295 | |||||||
| March 2005 | $ | 6.39 | 284,600 | 310,000 | January 2007 | March 2015 | 839 | 889 | |||||||||
| 636,452 | 661,852 | $ | 897 | $ | 1,184 | ||||||||||||
61
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
We have issued certain warrants to purchase shares of our common stock in
connection with equity financings pursuant to effective shelf registration
statements, and the holders of such warrants have the right to exercise them for
cash and to receive registered shares upon such exercise. In connection with the
issuance of these warrants, we agreed to file timely any reports required under
the Securities Exchange Act of 1934, as amended, to enable the delivery of
registered shares upon exercise of these warrants.
In March and July 2007, we amended certain warrant agreements to address
the presumption of net-cash settlement in the event that registered shares are
not available to settle the warrants. The amendments enable the settlement of
such warrants to be within our control. In particular, the amendments: (i)
preclude the warrant holders from exercising the warrants or require the warrant
holders to exercise the warrants on a net-share settled basis to enable the
issuance of shares that qualify for an exemption from registration under Section
3(a)(9) of the Securities Act of 1933, as amended, when there is no registration
statement in effect with respect to the shares underlying the warrants; (ii)
provide an explicit clarification that the warrants are not to be settled in
cash; and (iii) provide that we shall use reasonable best efforts to maintain
currently effective shelf registration statements, instead of requiring a
commitment to maintain the effectiveness of currently effective shelf
registration statements. On the effective date of these amendments, the change
in fair value from the most recent reporting date to the effective date of the
amendments was recorded in the consolidated statements of operations and the
then-current fair value for the warrants of $23,862,000 was reclassified from
liabilities to equity. Any changes in fair value subsequent to these
reclassifications shall not be recognized as long as the warrants continue to be
classified as equity. There were no reclassifications from liabilities to equity
for warrants in 2009 or 2008.
Non-employee options whose performance obligations are complete are
classified as derivative liabilities on our consolidated balance sheet. Upon the
exercise of these options, the instruments are marked to fair value and
reclassified from derivative liabilities to stockholders’ equity. In 2009,
reclassification of $130,000 from liabilities to equity has been included on our
consolidated balance sheet for such non-employee option exercises. In 2007, net
reclassification of $1,888,000 from equity to liabilities has been included on
our consolidated balance sheet to reflect completion of performance obligations
for certain non-employee options. No reclassifications were made in 2008 for
non-employee options.
Fair Value on a Recurring Basis
The following table presents information about our financial assets and
liabilities that are measured at fair value on a recurring basis as of December
31, 2009, and indicates the fair value category assigned.
| Fair Value Measurements at Reporting Date Using | ||||||||||||||||||
| Significant | ||||||||||||||||||
| Quoted Prices in | Other | Significant | ||||||||||||||||
| Active Markets for | Observable | Unobservable | ||||||||||||||||
| Identical Assets | Inputs | Inputs | ||||||||||||||||
| (In thousands) | Level 1 | Level 2 | Level 3 | Total | ||||||||||||||
| Assets | ||||||||||||||||||
| Money market funds (1) | $ | 33,395 | $ | — | $ | — | $ | 33,395 | ||||||||||
| U.S. Treasury securities (2) | 58,159 | — | — | 58,159 | ||||||||||||||
| Government-sponsored enterprise securities (2) | 14,021 | — | — | 14,021 | ||||||||||||||
| Corporate notes (2) | — | 59,498 | — | 59,498 | ||||||||||||||
| Total | $ | 105,575 | $ | 59,498 | $ | — | $ | 165,073 | ||||||||||
| Liabilities | ||||||||||||||||||
| Derivatives (3) | $ | — | $ | — | $ | 897 | $ | 897 | ||||||||||
| ____________________ | ||||||||||||||||||
| (1) | Included in cash and cash equivalents on our consolidated balance sheet. | |
| (2) | Included in marketable securities on our consolidated balance sheet. | |
| (3) | Included in fair value of derivatives on our consolidated balance sheet. | |
62
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Changes in Level 3 Recurring Fair Value
Measurements
The table below includes a rollforward of the balance sheet amounts for
the year ended December 31, 2009 (including the change in fair value), for
financial instruments in the Level 3 category. When a determination is made to
classify a financial instrument within Level 3, the determination is based upon
the significance of the unobservable parameters to the overall fair value
measurement. However, Level 3 financial instruments typically include, in
addition to the unobservable components, observable components (that is,
components that are actively quoted and can be validated to external sources).
Accordingly, the gains and losses in the table below include changes in fair
value due in part to observable factors that are part of the
methodology.
| Fair Value Measurements Using
Significant Unobservable Inputs (Level 3) Year Ended December 31, 2009 |
||||||||||||
| (In thousands) | Fair Value at December 31, 2008 |
Total Unrealized Gains Included in Earnings, net (1) |
Purchases, Sales, Issuances, Settlements, net |
Transfers In and/or Out of Level 3 |
Fair Value at December 31, 2009 |
Change in Unrealized Gains Related to Financial Instruments Held at December 31, 2009 (1) |
||||||
| Derivative liabilities | $1,184 | $(157) | $— | $(130) | $897 | $(215) | ||||||
| (1) | Reported as unrealized gain on fair value of derivatives in our consolidated statements of operations. |
Credit Risk
We place our cash, restricted cash, cash equivalents and marketable
securities with six financial institutions in the United States. Generally,
these deposits may be redeemed upon demand and therefore, bear minimal risk.
Deposits with banks may exceed the amount of insurance provided on such
deposits. Financial instruments that potentially subject us to concentrations of
credit risk consist primarily of marketable securities. Marketable securities
currently consist of investment grade U.S. Treasury securities, U.S.
government-sponsored enterprise securities and corporate notes. Our investment
policy, approved by the Board of Directors, limits the amount we may invest in
any one type of investment issuer, thereby reducing credit risk
concentrations.
3. JOINT VENTURE AND RELATED PARTY
TRANSACTIONS
TA Therapeutics, Ltd.
In March 2005, we and the Biotechnology Research Corporation (BRC), a
subsidiary of Hong Kong University of Science and Technology, established a
joint venture company in Hong Kong called TA Therapeutics, Ltd. (TAT). TAT
conducts research and was established to commercially develop products that
utilize telomerase activator drugs to restore the regenerative and functional
capacity of cells in various organ systems that have been impacted by
senescence, injury or chronic disease. On June 15, 2007, we and BRC entered into
an agreement to restructure the TAT joint venture. Under the amended agreements,
we direct the preclinical and drug development activities, own a 75% voting
interest and exercise control over the company. Upon any winding up of TAT, all
intellectual property of TAT is assigned to us and BRC is entitled to royalties
on sales of future products developed from TAT’s efforts up to a fixed amount
based on BRC’s cash contributions. Upon a winding up of TAT, if the assets
available for distribution, other than the intellectual property, are
insufficient to repay the whole of the paid-up capital, such assets shall be
distributed so that the losses shall be borne by the shareholders in proportion
to the cash contributed by both parties.
As a result of our obtaining control over TAT, we have included the
results of TAT in our consolidated financial statements beginning June 16, 2007.
Based on consideration of the relevant rights described above, we have
determined that BRC’s 25% equity interest in TAT is not substantive. The amended
arrangement represents, in substance, a research and development arrangement
between us and BRC. Therefore, this arrangement is being accounted for as a
research and development arrangement.
63
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Contributions from BRC
represent its share of funding for future research and development activities
that will be performed principally by BRC and partly by us. Accordingly, BRC’s
net contributions have been recorded as an advance payment for research and
development on our consolidated balance sheet. The advance payment from BRC has
been recognized as either a reduction of research and development expenses or
revenues from collaborative agreements depending upon who performs the related
research and development activity. The advance payment from BRC has been
recorded as a reduction of research and development expenses in our consolidated
statements of operations in the period when BRC performs the underlying research
activity on behalf of TAT. The advance payment from BRC has been recognized as
revenues from collaborative agreements in our consolidated statements of
operations in the period when we perform research activity on behalf of TAT and
the source of funds has not been derived from our cash contributions to TAT. For
the years ended December 31, 2009, 2008 and 2007, we recognized related party
revenue of none, $79,000 and $487,000, respectively. We incurred related party
research and development costs of $1,755,000, $794,000 and $941,000 for the
years ended December 31, 2009, 2008 and 2007, respectively. As of December 31,
2009 and 2008, the net balance of the advance payment from BRC was zero and
$440,000, respectively.
Start Licensing and ViaGen,
Inc.
In April 2005, Geron and Exeter Life Sciences, Inc. (Exeter) established
Start Licensing, Inc. (Start), a joint venture to manage and license a broad
portfolio of intellectual property rights related to animal reproductive
technologies. We and Exeter owned 49.9% and 50.1% of Start, respectively. In
connection with the establishment of Start, we granted a worldwide, exclusive,
non-transferable license to our patent rights to nuclear transfer technology for
use in animal cloning, with the right to sublicense such patent rights. Since
there was no net book value associated with the patent rights at the execution
of the joint venture, no initial value was recognized for our investment in
Start. We suspended the equity method of accounting since our proportionate
share of net losses in Start exceeded our original carrying value of the
investment and we had no commitments to provide financial support or obligations
to perform services or other activities for Start.
In August 2008, Geron and Exeter entered into Contribution Agreements
whereby we and Exeter exchanged our equity interests in Start for equity
interests in ViaGen, Inc. (ViaGen). As a result of the exchange, Start became a
wholly-owned subsidiary of ViaGen. Ownership of ViaGen immediately following the
transaction was as follows: Exeter – 69%; Geron – 27%; and Smithfield Foods –
4%. Since no value had been recorded for our investment in Start, the same zero
carrying value was applied to our investment in ViaGen. Geron’s share of equity
method losses from Start that were not recognized during the period the equity
method was suspended was carried over to the investment in ViaGen.
In September 2008, Geron provided a $1,500,000 loan to ViaGen in
connection with ViaGen’s acquisition of an interest in an unrelated company. The
loan bore an interest rate of 6% per annum and was convertible into ViaGen
equity at Geron’s option at the then current market value. Since the proceeds of
the loan did not fund prior ViaGen losses and represented additional financial
support to ViaGen, we applied the equity method of accounting to the basis of
the loan and recognized losses for our proportionate share of ViaGen’s operating
losses. The loan basis was reduced to zero as of March 31, 2009, and since we
had no commitments to provide financial support or obligations to perform
services or other activities for ViaGen, we suspended the equity method of
accounting.
In September 2009, Geron purchased $3,603,000 in equity from ViaGen and
simultaneously Exeter converted its outstanding debt with ViaGen into equity.
The new equity purchase did not fund prior ViaGen losses and represented
additional financial support to ViaGen. Ownership of ViaGen upon consummation of
the transactions and at December 31, 2009 was as follows: Exeter – 70%; Geron –
28%; and Smithfield Foods – 2%. Subsequent to our equity purchase, Geron
received $1,593,000 from ViaGen in repayment of the 2008 loan, including accrued
interest. As the source of funds to repay the loan and accrued interest was
derived from our equity purchase, the equity investment in ViaGen was recorded
net of the loan and interest payment. We have no commitments to provide
financial support or obligations to perform services or other activities for
ViaGen.
64
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
With the new investment in 2009, we resumed applying the equity method of
accounting by increasing (decreasing) the carrying value of our investment by
our proportionate share of ViaGen’s earnings (losses). If equity method losses
exceed the carrying value of the investment, losses will be applied against any
advances to ViaGen, including any commitments to provide financial support until
those amounts are reduced to zero. The equity method of accounting shall then be
suspended until income is subsequently reported. If income is reported, Geron’s
proportionate share of income shall first be applied to recognize the equity
method losses accumulated during the time the equity method was
suspended.
For the years ended December 31, 2009 and 2008, we recognized $1,338,000
and $844,000, respectively, for our proportionate share of ViaGen’s operating
losses. Our share of losses is recorded in the consolidated statements of
operations under losses recognized under equity method investment. The adjusted
basis of our investment in ViaGen at December 31, 2009 and 2008 was $1,328,000
and $656,000, respectively, which is reflected under investments in licensees on
our consolidated balance sheet.
4. PROPERTY AND EQUIPMENT
Property and equipment, stated at
cost, is comprised of the following:
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| (In thousands) | ||||||||
| Furniture and computer equipment | $ | 4,298 | $ | 4,232 | ||||
| Lab equipment | 10,616 | 10,124 | ||||||
| Leasehold improvements | 7,497 | 7,315 | ||||||
| 22,411 | 21,671 | |||||||
| Less accumulated depreciation and amortization | (18,473 | ) | (17,285 | ) | ||||
| $ | 3,938 | $ | 4,386 | |||||
5. EQUIPMENT LINE
In 2009, we renewed our equipment financing facility and had
approximately $500,000 available for borrowing as of December 31, 2009. This
facility is secured by a certificate of deposit. Any outstanding principal
balance bears a fixed interest rate equal to one and one-half percentage point
above the Prime Rate. No amounts were due under this facility as of December 31,
2009 and 2008.
6. ACCRUED LIABILITIES
Accrued liabilities consist of the
following:
| December 31, | ||||||||||
| 2009 | 2008 | |||||||||
| (In thousands) | ||||||||||
| Sponsored research agreements | $ | 107 | $ | 178 | ||||||
| Service provider obligations | 274 | 237 | ||||||||
| Clinical trials | 698 | 649 | ||||||||
| Related party payable | — | 270 | ||||||||
| Other | 846 | 914 | ||||||||
| $ | 1,925 | $ | 2,248 | |||||||
7. COMMITMENTS AND CONTINGENCIES
Operating Lease Commitment
In March 2008, as payment of the total rent due for our premises at 200
Constitution Drive and 230 Constitution Drive in Menlo Park, California, for the
period from August 1, 2008 through July 31, 2012, we issued to the lessor of
those premises 742,158 shares of our common stock. The fair value of the common
stock of $3,191,000 was recorded as a prepaid asset and is being amortized to
rent expense on a straight-line basis over the lease period.
65
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
In May 2007, as payment of the total rent due for our premises at 149
Commonwealth Drive in Menlo Park, California, for the period from May 1, 2007
through April 30, 2010, we issued 210,569 shares of our common stock to the
lessor of those premises. The fair value of the common stock of $1,573,000 was
recorded as a prepaid asset and is being amortized to rent expense on a
straight-line basis over the lease period.
Future minimum payments under non-cancelable operating leases are zero
through July 31, 2012, as a result of the prepayments of rent with our common
stock. Rent expense under operating leases was approximately $1,324,000,
$1,259,000 and $1,029,000 for the years ended December 31, 2009, 2008 and 2007,
respectively.
Severance Plan
We have a Change of Control Severance Plan (the Severance Plan) that
applies to all employees, and provides for each employee to receive a severance
payment upon a triggering event following a change of control. A triggering
event is defined as an event where: (i) an employee is terminated by us without
cause in connection with a change of control or within 12 months following a
change of control; or (ii) an employee is not offered comparable employment (new
or continuing) by us or our successor or acquirer within 30 days after the
change of control or any employment offer is rejected; or (iii) after accepting
(or continuing) employment with us after a change of control, an employee
resigns within six months following a change of control due to a material change
in the terms of employment. Severance payments range from two to 18 months of
base salary, depending on the employee’s position with us, payable in a lump sum
payment. We have not made any payments under our Severance Plan.
Indemnifications to Officers and
Directors
Our corporate bylaws require that we indemnify our officers and
directors, as well as those who act as directors and officers of other entities
at our request, against expenses, judgments, fines, settlements and other
amounts actually and reasonably incurred in connection with any proceedings
arising out of their services to Geron. In addition, we have entered into separate indemnification agreements
with each of our directors which provide for indemnification of these directors
under similar circumstances and under additional circumstances. The
indemnification obligations are more fully described in our bylaws and the
indemnification agreements. We purchase standard insurance to cover claims or a
portion of the claims made against our directors and officers. Since a maximum
obligation is not explicitly stated in our bylaws or in our indemnification
agreements and will depend on the facts and circumstances that arise out of any
future claims, the overall maximum amount of the obligations cannot be
reasonably estimated. Historically, we have not made payments related to these
obligations, and the fair value of these obligations was zero on our
consolidated balance sheets as of December 31, 2009 and 2008.
8. STOCKHOLDERS’ EQUITY
Warrants
As of December 31, 2009, the following warrants to purchase our common
stock were outstanding and classified as equity:
| Issuance Date | Exercise Price | Number of Shares | Exercisable Date | Expiration Date | |||||||
| September 2009 | $ | 9.00 | 150,000 | September 2009 | September 2014 | ||||||
| October 2007 | $ | 7.42 | 25,000 | October 2007 | October 2012 | ||||||
| September 2007 | $ | 7.19 | 100,000 | September 2007 | September 2012 | ||||||
| February 2007 | $ | 6.80 | 1,125,000 | June 2007 | December 2011 | ||||||
| December 2006 | $ | 6.80 | 3,000,000 | June 2007 | December 2011 | ||||||
| April 2005 | $ | 3.75 | 470,000 | April 2005 | April 2015 | ||||||
| November 2004 | $ | 6.80 | 2,295,082 | May 2005 | November 2011 | ||||||
| September 2001 | $ | 9.07 | 5,000 | September 2001 | September 2011 | ||||||
| August 2001 | $ | 14.60 | 100,000 | August 2001 | August 2011 | ||||||
| August 2000 | $ | 31.69 | 5,000 | August 2000 | August 2010 | ||||||
| July 2000 | $ | 6.75 | 25,000 | July 2000 | July 2010 | ||||||
| March 2000 | $ | 17.50 | 200,000 | March 2000 | March 2012 | ||||||
| March 2000 | $ | 12.50 | 100,000 | March 2000 | March 2012 | ||||||
| 7,600,082 | |||||||||||
66
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
In April 2009 in connection with our continued collaboration with an
investor and licensee and the data received under the collaboration relevant to
Geron’s therapeutic programs, we modified the terms of certain outstanding
warrants held by this investor by extending the exercise term and reducing the
exercise price. The exercise term of warrants to purchase 200,000 shares of
common stock was extended to March 9, 2012 from March 9, 2010 and the exercise
price was modified to $17.50 per share from $67.09 per share. The exercise term
of warrants to purchase 100,000 shares of common stock was extended to March 9,
2012 from March 9, 2010 and the exercise price was unchanged at $12.50 per
share. In connection with the modifications, we recognized a deemed dividend of
approximately $190,000 in our consolidated statements of operations for the
incremental fair value of the modified warrants, as calculated using the Black
Scholes option-pricing model as of the modification date.
In February 2007 in exchange for the exercise of warrants to purchase
1,875,000 shares of common stock, we issued warrants to purchase 1,125,000
shares of common stock, at a premium, exercisable from June 2007. The new
warrants (2007 D Warrants) were substantially the same as the 2006 A Warrants
issued in the December 2006 financing and were issued to the same institutional
investors who held the 2006 A Warrants. The aggregate fair value of $3,661,000
for the 2007 D Warrants, as calculated using the Black Scholes option-pricing
model, was recognized as a deemed dividend in our consolidated statements of
operations.
In December 2007, we modified the
terms of certain outstanding warrants by extending the exercise term and
reducing the exercise price. The exercise term of the 2004 A Warrants to
purchase 2,295,082 shares of common stock was extended to November 2011 and the
exercise price was modified to $7.50 per share. The exercise terms of the 2006 A
Warrants to purchase 3,000,000 shares of common stock and 2007 D Warrants to
purchase 1,125,000 shares of common stock were extended to December 2011 and the
exercise prices were modified to $7.50 per share. In connection with the
modifications, we received $3,630,000 in cash consideration from the
institutional investors holding the outstanding warrants. We recognized a deemed
dividend of $5,420,000 in our consolidated statements of operations for the
incremental fair value of the modified warrants, as calculated using the Black
Scholes option-pricing model as of the modification date, net of the cash
consideration received from the institutional investors for the modifications.
As of December 15, 2009, the exercise price for each of these warrants was reset
to $6.80 per share in accordance with the terms of the modified warrant
agreements.
1992 Stock Option Plan
The 1992 Stock Option Plan (1992
Plan) expired in August 2002 and no further option grants can be made from the
1992 Plan. The options granted under the 1992 Plan were either incentive stock
options or nonstatutory stock options. Options granted under the 1992 Plan
expired no later than ten years from the date of grant. For incentive stock
options and nonstatutory stock options, the option exercise price was at least
100% and 85%, respectively, of the fair market value of the underlying common
stock on the date of grant. Options to purchase shares of common stock generally
vested over a period of four or five years from the date of the option grant,
with a portion vesting after six months and the remainder vesting ratably over
the remaining period.
2002 Equity Incentive Plan
In May 2002, our stockholders approved the adoption of the 2002 Equity
Incentive Plan (2002 Plan) to replace the 1992 Plan. Our Board of Directors
administers the 2002 Plan. The 2002 Plan provides for grants to employees of us
or of our subsidiary (including officers and employee directors) of either
incentive stock or nonstatutory stock options and stock purchase rights to
employees (including officers and employee directors) and consultants (including
non-employee directors) of us or of our subsidiary. As of December 31, 2009, we
had reserved 17,579,603 shares of common stock for issuance under the 2002 Plan.
Options granted under the 2002 Plan expire no later than ten years from the date
of grant. For incentive stock options, the exercise price shall be equal to 100%
of the fair market value of the underlying common stock on the date of grant.
Exercise prices for all other stock options are determined by the administrator.
If, at the time we grant an option, the optionee directly or by attribution owns
stock possessing more than 10% of the total combined voting power of all classes
of our stock, the option price shall be at least 110% of the fair market value
of the underlying common stock and shall not be exercisable more than five years
after the date of grant.
Options to purchase shares of common stock generally vest over a period
of four years from the date of the option grant, with a portion vesting after
six months and the remainder vesting ratably over the remaining period. Stock
purchase rights (restricted stock awards and restricted stock units) have
variable vesting schedules and purchase prices as determined by the Board of
Directors on the date of grant.
67
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Under certain circumstances, options may be exercised prior to vesting,
subject to our right to repurchase shares subject to such option at the exercise
price paid per share. Our repurchase rights would generally terminate on a
vesting schedule identical to the vesting schedule of the exercised option. In
2009 and 2008, we repurchased none and 114,914 shares, respectively, related to
restricted stock awards for payroll tax withholdings. As of December 31, 2009,
no shares outstanding were subject to repurchase.
1996 Directors’ Stock Option Plan
The 1996 Directors’ Stock Option Plan (1996 Directors Plan) expired in
July 2006 and no further option grants can be made from the 1996 Directors Plan.
The options granted under the 1996 Directors Plan were nonstatutory stock
options and expired no later than ten years from the date of grant. The option
exercise price was equal to the fair market value of the underlying common stock
on the date of grant. Options to purchase shares of common stock generally were
100% vested upon grant, except for options granted upon first appointment to the
Board of Directors (First Option). The First Option vested annually over three
years upon each anniversary date of appointment to the Board. The options issued
pursuant to the 1996 Directors Plan remain exercisable for up to 90 days
following the optionee’s termination of service as our director, unless such
termination is a result of death or permanent and total disability, in which
case the options (both those already exercisable and those that would have
become exercisable had the director remained on the Board of Directors for an
additional 36 months) remain exercisable for up to a 24 month period.
2006 Directors’ Stock Option Plan
In May 2006, our stockholders approved the adoption of the 2006
Directors’ Stock Option Plan (2006 Directors Plan) to replace the 1996 Directors
Plan. As of December 31, 2009, we had reserved an aggregate of 2,500,000 shares
of common stock for issuance under the 2006 Directors Plan. The 2006 Directors
Plan provides for the automatic grant of the following types of equity
awards.
First Option. Each
person who becomes a non-employee director, whether by election of the
stockholders of the Company or by appointment by the Board of Directors to fill
a vacancy, will automatically be granted an option to purchase 45,000 shares of
common stock on the date on such person first becomes a non-employee director
(the First Option).
Subsequent Awards. Each non-employee director (other than the Chairman of the Board of
Directors and any director receiving a First Option on the date of the annual
meeting) will automatically be granted a subsequent option on the date of the
Annual Meeting of Stockholders in each year during such director’s service on
the Board (a Subsequent Option) to purchase 10,000 shares of common stock and a
restricted stock award (a Subsequent Stock Award) of 5,000 shares of common
stock. In the case of the Chairman of the Board, the Subsequent Option will be
for 20,000 shares of common stock and the Subsequent Stock Award shall be for
10,000 shares of common stock.
Committee Chair Service Awards. On the date of each Annual Meeting of
Stockholders, the Chairman of the Audit Committee receives an option to purchase
5,000 shares of common stock (a Committee Chair Service Option), and a
restricted stock award (a Committee Chair Service Stock Award) of 2,500 shares
of common stock. The Committee Chair Service Option for the Compensation
Committee Chairman and the Nominating Committee Chairman shall be for 2,500
shares of common stock and the Committee Chair Service Stock Award shall be for
1,250 shares of common stock.
Committee Service Awards.
Upon each non-employee
director’s appointment to the Audit Committee, Compensation Committee or Nominating Committee of the Board of
Directors, the director will receive an option to purchase 2,500 shares of
common stock (a First Committee Service Option). Thereafter, an option to
purchase 1,250 shares of common stock (a Subsequent Committee Service Option)
and a restricted stock award of 625 shares of common stock (a Subsequent
Committee Service Stock Award) shall be granted to each non-employee director on
the date of each Annual Meeting during the director’s service on such committee,
other than the Chairman of such committee. There is currently no stock option
grant or restricted stock award contemplated for participation on other
committees.
The 2006 Directors Plan provides that each First Option vests annually
over three years upon each anniversary date of appointment to the Board. Each
Subsequent Option, Committee Chair Service Option, First Committee Service
Option and Subsequent Committee Service Option is fully vested
68
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
on the date of its
grant. Each Subsequent Stock Award, Committee Chair Service Stock Award and
Subsequent Committee Service Stock Award vests annually in four equal
installments over four years commencing on the date of grant and no payment
shall be required from the non-employee director in order to receive the award.
Options under the 2006 Directors Plan remain exercisable for up to 90 days
following the optionee’s termination of service as our director, unless such
termination is a result of death or permanent and total disability, in which
case the options (both those already exercisable and those that would have
become exercisable had the director remained on the Board of Directors for an
additional 36 months) remain exercisable for up to a 24 month period or unless
there is a death of an optionee within 3 months following his or her termination
of service, in which case the options will remain exercisable for an additional
six month period from the date of death. Upon termination of service as our
director, any unvested options and restricted stock awards shall return to the
2006 Directors Plan, unless such termination is a result of death or permanent
and total disability, in which case any unvested options and restricted stock
awards shall immediately vest.
The exercise price of all options granted under the 2006 Directors Plan
is equal to 100% of the fair market value of the underlying common stock on the
date of grant. Options granted under the 2006 Directors Plan have a term of ten
years.
Aggregate option activity for the 1992 Plan, 2002 Plan, 1996 Directors
Plan and 2006 Directors Plan is as follows:
| Outstanding Options | ||||||||||||||||||||
| Weighted Average | Aggregate | |||||||||||||||||||
| Shares | Weighted Average | Remaining | Intrinsic | |||||||||||||||||
| Available | Number of | Exercise Price | Contractual Life | Value | ||||||||||||||||
| For Grant | Shares | Per Share | (In years) | (In thousands) | ||||||||||||||||
| Balance at December 31, 2006 | 7,726,616 | 9,006,446 | $ | 7.77 | $ | 18,290 | ||||||||||||||
| Additional shares authorized | 2,000,000 | — | $ | — | ||||||||||||||||
| Options granted | (1,674,759 | ) | 1,674,759 | $ | 8.29 | |||||||||||||||
| Awards granted | (2,170,882 | ) | — | $ | — | |||||||||||||||
| Options exercised | — | (282,597 | ) | $ | 5.95 | |||||||||||||||
| Options canceled/forfeited | 387,872 | (387,872 | ) | $ | 9.15 | |||||||||||||||
| Awards canceled/repurchased | 19,900 | — | $ | — | ||||||||||||||||
| 1992 Plan and 1996 Directors | ||||||||||||||||||||
| Plan options expired | (129,892 | ) | — | $ | 11.75 | |||||||||||||||
| Balance at December 31, 2007 | 6,158,855 | 10,010,736 | $ | 7.86 | $ | 2,596 | ||||||||||||||
| Additional shares authorized | 2,000,000 | — | $ | — | ||||||||||||||||
| Options granted | (2,060,025 | ) | 2,060,025 | $ | 3.99 | |||||||||||||||
| Awards granted | (1,227,522 | ) | — | $ | — | |||||||||||||||
| Options exercised | — | (146 | ) | $ | 3.97 | |||||||||||||||
| Options canceled/forfeited | 1,584,685 | (1,584,685 | ) | $ | 6.19 | |||||||||||||||
| Awards canceled/repurchased | 209,929 | — | $ | — | ||||||||||||||||
| 1992 Plan and 1996 Directors | ||||||||||||||||||||
| Plan options expired | (844,474 | ) | — | $ | 5.54 | |||||||||||||||
| Balance at December 31, 2008 | 5,821,448 | 10,485,930 | $ | 7.35 | $ | 2,071 | ||||||||||||||
| Additional shares authorized | 2,000,000 | — | $ | — | ||||||||||||||||
| Options granted | (2,767,879 | ) | 2,767,879 | $ | 6.49 | |||||||||||||||
| Awards granted | (1,916,772 | ) | — | $ | — | |||||||||||||||
| Options exercised | — | (320,876 | ) | $ | 5.59 | |||||||||||||||
| Options canceled/forfeited | 1,171,538 | (1,171,538 | ) | $ | 10.03 | |||||||||||||||
| Awards canceled/repurchased | 73,069 | — | $ | — | ||||||||||||||||
| 1992 Plan and 1996 Directors | ||||||||||||||||||||
| Plan options expired | (913,967 | ) | — | $ | 10.97 | |||||||||||||||
| Balance at December 31, 2009 | 3,467,437 | 11,761,395 | $ | 6.93 | 6.37 | $ | 4,553 | |||||||||||||
| Options exercisable at | ||||||||||||||||||||
| December 31, 2009 | 8,003,110 | $ | 7.33 | 5.21 | $ | 3,010 | ||||||||||||||
| Options fully vested and expected | ||||||||||||||||||||
| to vest at December 31, 2009 | 11,318,051 | $ | 6.96 | 6.27 | $ | 4,398 | ||||||||||||||
69
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The aggregate intrinsic value in the preceding table represents the total
intrinsic value, based on Geron’s closing stock price of $5.55 per share as of
December 31, 2009, which would have been received by the option holders had all
the option holders exercised their options as of that date.
There were no options granted with an exercise price below fair market
value of our common stock on the date of grant for 2009, 2008 and 2007. There
were no options granted with an exercise price greater than grant date fair
market value in 2009 or 2008. There were 6,000 options granted to employees with
an exercise price greater than grant date fair market value with a weighted
average exercise price of $7.48 per share in 2007. As of December 31, 2009, 2008
and 2007, there were 8,003,110, 7,483,714 and 7,308,554 exercisable options
outstanding at weighted average exercise prices per share of $7.33, $8.05 and
$7.98, respectively.
The total pretax intrinsic value of stock options exercised during 2009,
2008 and 2007 was $747,000, none and $741,000, respectively. Cash received from
the exercise of options in 2009, 2008 and 2007 totaled approximately $1,793,000,
$1,000 and $1,681,000, respectively. No income tax benefit was realized from
stock options exercised in 2009 since we reported an operating
loss.
Information about stock options
outstanding as of December 31, 2009 is as follows:
| Options Outstanding | |||||||||||||||
| Weighted Average | |||||||||||||||
| Weighted Average | Remaining | ||||||||||||||
| Number of | Exercise Price | Contractual Life | |||||||||||||
| Exercise Price Range | Shares | Per Share | (In years) | ||||||||||||
| $ | 1.83 | –$ | 4.97 | 2,607,481 | $ | 3.91 | 6.86 | ||||||||
| $ | 4.97 | –$ | 6.40 | 2,089,092 | $ | 5.96 | 5.29 | ||||||||
| $ | 6.40 | –$ | 6.95 | 3,514,739 | $ | 6.58 | 8.44 | ||||||||
| $ | 6.95 | –$ | 41.13 | 3,550,083 | $ | 10.06 | 4.61 | ||||||||
| $ | 1.83 | –$ | 41.13 | 11,761,395 | $ | 6.93 | 6.37 | ||||||||
Aggregate restricted stock activity
for the 2002 Plan and 2006 Directors Plan is as follows:
| Weighted Average | Weighted Average | |||||||||||
| Grant Date | Remaining | |||||||||||
| Number of | Fair Value | Contractual Term | ||||||||||
| Shares | Per Share | (In years) | ||||||||||
| Non-vested restricted stock at December 31, 2006 | 40,000 | $ | 7.57 | 1.54 | ||||||||
| Granted | 2,170,882 | $ | 8.64 | — | ||||||||
| Vested | (642,903 | ) | $ | 7.49 | — | |||||||
| Canceled/forfeited | (16,325 | ) | $ | 9.32 | — | |||||||
| Non-vested restricted stock at December 31, 2007 | 1,551,654 | $ | 9.08 | 1.05 | ||||||||
| Granted | 1,227,522 | $ | 4.21 | — | ||||||||
| Vested | (1,427,626 | ) | $ | 6.54 | — | |||||||
| Canceled/forfeited | (95,015 | ) | $ | 7.63 | — | |||||||
| Non-vested restricted stock at December 31, 2008 | 1,256,535 | $ | 7.32 | 2.42 | ||||||||
| Granted | 1,916,772 | $ | 6.14 | — | ||||||||
| Vested | (1,427,654 | ) | $ | 7.11 | — | |||||||
| Canceled/forfeited | (73,069 | ) | $ | 6.11 | — | |||||||
| Non-vested restricted stock at December 31, 2009 | 1,672,584 | $ | 6.20 | 3.32 | ||||||||
The total fair value of restricted stock that vested during 2009, 2008
and 2007 was $8,633,000, $6,184,000 and $4,820,000, respectively.
Employee Stock Purchase Plan
In July 1996, we adopted the 1996 Employee Stock Purchase Plan (Purchase
Plan) and as of December 31, 2009, we had reserved an aggregate of 1,200,000
shares of common stock for issuance under the Purchase Plan. Approximately
572,000 and 479,000 shares have been issued under the Purchase Plan as of
December 31, 2009 and 2008, respectively. As of December 31, 2009, 627,951
shares were available for issuance under the Purchase Plan.
70
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Under the terms of the Purchase Plan, employees can choose to have up to
10% of their annual salary withheld to purchase our common stock. An employee
may not make additional payments into such account or increase the withholding
percentage during the offering period.
The Purchase Plan is comprised of a series of offering periods, each with
a maximum duration (not to exceed 12 months) with new offering periods
commencing on January 1 and July 1 of each year. The date an employee enters the
offering period will be designated his or her entry date for purposes of that
offering period. An employee may only participate in one offering period at a
time. Each offering period consists of two consecutive purchase periods of six
months’ duration, with the last day of such period designated a purchase
date.
The purchase price per share at which common stock is purchased by the
employee on each purchase date within the offering period is equal to 85% of the
lower of (i) the fair market value per share of Geron common stock on the
employee’s entry date into that offering period or (ii) the fair market value
per share of common stock on that purchase date. If the fair market value of
Geron common stock on the purchase date is less than the fair market value at
the beginning of the offering period, a new 12 month offering period will
automatically begin on the first business day following the purchase date with a
new fair market value.
Effective for the offering period beginning July 1, 2009 and subsequent
offering periods, shares purchased under the Purchase Plan shall be registered
and available for trading in an open market transaction one year from the date
of purchase, and certificates evidencing such shares shall bear a restrictive
legend.
Stock-Based Compensation
Expense
We measure and recognize compensation expense for all share-based payment
awards made to employees and directors, including employee stock options,
restricted stock awards and employee stock purchases related to the Purchase
Plan, based on estimated grant-date fair values.
The following table summarizes the stock-based compensation expense
related to share-based payment awards for the years ended December 31, 2009,
2008 and 2007 which was allocated as follows:
| Year Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| (In thousands) | |||||||||
| Research and development | $ | 5,339 | $ | 5,492 | $ | 6,064 | |||
| General and administrative | 5,236 | 6,001 | 5,303 | ||||||
| Stock-based compensation expense included in operating expenses | $ | 10,575 | $ | 11,493 | $ | 11,367 | |||
The fair value of options granted in fiscal years 2009, 2008 and 2007
reported above has been estimated at the date of grant using the Black Scholes
option-pricing model with the following assumptions:
| 2009 | 2008 | 2007 | ||||
| Dividend yield | 0% | 0% | 0% | |||
| Expected volatility range | 0.630 to 0.633 | 0.527 to 0.596 | 0.737 to 0.774 | |||
| Risk-free interest rate range | 1.54% to 2.52% | 2.08% to 3.57% | 3.40% to 5.05% | |||
| Expected term | 5 yrs | 5 yrs | 5 yrs |
The fair value of employee stock purchases in fiscal years 2009, 2008 and
2007 under the Purchase Plan has been estimated using the Black Scholes
option-pricing model with the following assumptions:
| 2009 | 2008 | 2007 | |||||
| Dividend yield | 0% | 0% | 0% | ||||
| Expected volatility range | 0.536 to 1.016 | 0.458 to 0.593 | 0.419 to 0.471 | ||||
| Risk-free interest rate range | 0.28% to 2.38% | 2.13% to 4.97% | 4.97% to 5.26% | ||||
| Expected term | 6 mos to 12 mos | 6 mos to 12 mos | 6 mos to 12 mos |
71
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Dividend yield is based on historical cash dividend payments, which have
been none to date. Expected volatility range is based on historical volatilities
of our stock since traded options on Geron stock do not correspond to option
terms and trading volume of options is limited. The risk-free interest rate
range is based on the U.S. Zero Coupon Treasury Strip Yields for the expected
term in effect on the date of grant for an award. The expected term of options
is derived from actual historical exercise data and represents the period of
time that options granted are expected to be outstanding. The expected term of
employees’ purchase rights under the Purchase Plan is equal to the purchase
period. We grant options under our equity plans to employees, non-employee
directors, and consultants for whom the vesting period is generally four
years.
As stock-based compensation expense recognized in the consolidated
statements of operations for the years ended December 31, 2009, 2008 and 2007 is
based on awards ultimately expected to vest, it has been reduced for estimated
forfeitures but at a minimum, reflects the grant-date fair value of those awards
that actually vested in the period. Forfeitures have been estimated at the time
of grant and revised, if necessary, in subsequent periods if actual forfeitures
differ from those estimates. Forfeitures were estimated based on historical
experience.
Based on the Black Scholes option-pricing model, the weighted average
estimated fair value of employee stock options granted during the years ended
December 31, 2009, 2008 and 2007 was $3.55, $2.06 and $5.37 per share,
respectively. The weighted average estimated fair value of purchase rights under
our Purchase Plan for the years ended December 31, 2009, 2008 and 2007 was
$3.17, $1.40 and $2.34 per share, respectively. As of December 31, 2009, total
compensation cost related to unvested stock awards not yet recognized was
$17,583,000, net of estimated forfeitures, which is expected to be recognized
over the next 41 months on a weighted-average basis.
Stock-Based Compensation to Service
Providers
We grant options, restricted stock
and warrants to purchase common stock to consultants from time to time in
exchange for services performed for us. In general, the options and restricted
stock vest over the contractual period of the consulting arrangement and
warrants are fully vested on the grant date. No options or warrants were granted
to consultants in 2009 or 2008. We granted options and warrants to consultants
to purchase 125,000 shares of our common stock in 2007. In September 2009, our
Chief Scientific Officer for Telomerase Technologies retired and became an
advisor to the Company. In connection with his advisory function, the options
and restricted stock awards previously granted to him as an employee continue to
vest under the same schedule as he provides services to the Company, and such
awards are accounted for as consultant awards. The fair value of options,
restricted stock awards and warrants granted to consultants is being amortized
to expense over the vesting term of the respective equity award. In addition, we
will record any additional increase in the fair value of the options, restricted
stock awards or warrants as the respective equity award vests. We recorded
stock-based compensation expense of $190,000, none and $1,466,000 for the vested
portion of the fair value of options, restricted stock awards and warrants to
consultants in 2009, 2008 and 2007, respectively.
We also grant common stock to
consultants, vendors and research institutions in exchange for services either
performed or to be performed for us. In 2009, 2008 and 2007, we issued
1,272,438, 2,294,685 and 1,169,823 shares of common stock, respectively, in
exchange for goods or services. For these stock grants, we record a prepaid
asset equal to the fair market value of the granted shares on the date of grant
and amortize to expense on a pro-rata basis as services are performed or goods
are received. In 2009, 2008 and 2007, we recognized approximately $7,230,000,
$8,723,000 and $6,304,000, respectively, of expense in connection with stock
grants to consultants, vendors and research institutions. As of December 31,
2009, $3,109,000 related to vendor stock grants remained as a prepaid asset
which is being amortized to research and development expense on a pro-rata basis
as services are incurred or goods are received. Also, we have prepaid our rental
obligation for our facilities with common stock and as of December 31, 2009,
have a prepaid balance of $2,236,000 which is being amortized to rent expense on
a straight-line basis over the term of the leases until July 31,
2012.
72
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Common Stock Reserved for Future Issuance
Common stock reserved for future issuance as of December 31, 2009 is as
follows:
| Outstanding stock options | 11,761,395 |
| Options and awards available for grant | 3,467,437 |
| Employee stock purchase plan | 627,951 |
| Warrants outstanding | 7,951,934 |
| Total | 23,808,717 |
Share Purchase Rights Plan
On July 20, 2001, our Board of Directors adopted a share purchase rights
plan and declared a dividend distribution of one right for each outstanding
share of common stock to stockholders of record as of July 31, 2001. Each right
entitles the holder to purchase one unit consisting of one one-thousandth of a
share of Series A Junior Participating Preferred Stock for $100 per unit. Under
certain circumstances, if a person or group acquires 15% or more of our
outstanding common stock, holders of the rights (other than the person or group
triggering their exercise) will be able to purchase, in exchange for the $100
exercise price, shares of our common stock, par value $0.001 per share, or of
any company into which we are merged having a value of $200. The rights expire
on July 31, 2011 unless extended by our Board of Directors. As of December 31,
2009, no rights were exercisable into any shares of common stock.
401(k) Plan
We sponsor a defined-contribution savings plan under Section 401(k) of
the Internal Revenue Code covering all full-time U.S. employees (Geron 401K
Plan). Participating employees may contribute up to the annual Internal Revenue
Service contribution limit. The Geron 401K Plan also permits us to provide
discretionary matching and profit sharing contributions. The Geron 401K Plan is
intended to qualify under Section 401 of the Internal Revenue Code so that
contributions by employees or by us, and income earned on the contributions, are
not taxable to employees until withdrawn from the Geron 401K Plan. Our
contributions, if any, will be deductible by us when made. At the direction of
each participant, the assets of the Geron 401K Plan are invested in any of 14
different investment options.
In December 2009, 2008 and 2007, our Board of Directors approved a
matching contribution equal to 100% of each employee’s 2009, 2008 and 2007
contributions, respectively. The matching contributions are invested in our
common stock and vest ratably over four years for each year of service completed
by the employee, commencing from the date of hire, until it is fully vested when
the employee has completed four years of service. We provided the matching
contribution in the month following Board approval.
For the vested portion of the 2009 match under this plan, we recorded
$790,000 as research and development expense and $182,000 as general and
administrative expense. For the vested portion of the 2008 match under this
plan, we recorded $631,000 as research and development expense and $134,000 as
general and administrative expense. For the vested portion of the 2007 match
under this plan, we recorded $570,000 as research and development expense and
$70,000 as general and administrative expense. As of December 31, 2009,
approximately $517,000 remained unvested for the 2008, 2007 and 2006
matches.
Public Offering
On February 19, 2009, we completed an underwritten public offering of
7,250,000 shares of our common stock at a public offering price of $6.60 per
share, resulting in net cash proceeds of approximately $45,933,000 after
deducting underwriting discounts and commissions and offering
expenses.
Offering of Common Stock and
Warrants
In September 2009, we sold 550,000 shares of Geron common stock (Shares)
and warrants to purchase an additional 150,000 shares of common stock with an
exercise price of $9.00 per share (Warrants) to certain institutional investors
for total gross proceeds of $3,603,000. The Shares, Warrants and shares of
common stock underlying the warrants (Warrant Shares) were issued through a
prospectus supplement to an effective shelf
73
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
registration statement.
The Warrants are immediately exercisable for a period of five years from the
date of issuance. A lock-up agreement limits the sale or other disposition of
the Shares, the Warrants and the Warrant Shares by the investors for a period of
one year beginning on September 9, 2009.
9. COLLABORATIVE AGREEMENT
In June 2009, we entered into a worldwide exclusive license and alliance
agreement with GE Healthcare UK, Limited (GEHC) to develop and commercialize
cellular assay products derived from human embryonic stem cells (hESCs) for use
in drug discovery, development and toxicity screening. Under the terms of the
agreement, GEHC has been granted an exclusive license under Geron’s intellectual
property portfolio covering the growth and differentiation of hESCs, as well as
a sublicense under Geron’s rights to the hESC patents held by the Wisconsin
Alumni Research Foundation. We have established a multi-year alliance program
with GEHC under which scientists from both companies will work to develop
hESC-based products for drug discovery.
In connection with the agreement, we received upfront non-refundable
license payments under the exclusive license and sublicense and can receive
milestone payments upon achievement of certain commercial development and
product sales events and royalties on future product sales. Under the alliance
program, GEHC is responsible for all costs incurred by GEHC and all costs
incurred by Geron for activities undertaken at Geron, including the funding of
Geron scientists working on the alliance program. An Alliance Steering
Committee, with representatives from each company, coordinates and manages the
alliance program.
License payments under the GEHC agreement were recorded as deferred
revenue upon receipt and are being recognized ratably as revenue over the
alliance program period as a result of our continuing involvement with the
collaboration. Funding received for Geron’s efforts under the alliance program
is being recognized as revenue as costs are incurred, which approximates our
level of effort over the period of the alliance program. Since the milestone
payments are subject to substantive contingencies, any such payments will be
recognized upon completion of the specified milestones. Royalties received under
the agreement will generally be recognized as revenue upon receipt of the
related royalty payment. For the year ended December 31, 2009, we recognized
$450,000 as revenues from collaborative agreements and $350,000 in license fee
revenue in connection with this agreement.
10. INCOME TAXES
Deferred income taxes reflect the net tax effects of temporary
differences between the carrying amounts of assets and liabilities for financial
reporting purposes and the amounts used for income tax purposes. Significant
components of our deferred tax assets as of December 31 are as follows:
| 2009 | 2008 | ||||||||
| (In thousands) | |||||||||
| Net operating loss carryforwards | $ | 179,800 | $ | 157,600 | |||||
| Purchased technology | 11,400 | 12,600 | |||||||
| Research credits | 21,900 | 22,600 | |||||||
| Capitalized research and development | 15,600 | 14,600 | |||||||
| License fees | 1,900 | 2,200 | |||||||
| Other — net | 8,800 | 10,900 | |||||||
| Total deferred tax assets | 239,400 | 220,500 | |||||||
| Valuation allowance for deferred tax assets | (239,400 | ) | (220,500 | ) | |||||
| Net deferred tax assets | $ | — | $ | — | |||||
We record net deferred tax assets to the extent we believe these assets
will more likely than not be realized. In making such determination, we consider
all available positive and negative evidence, including scheduled reversals of
deferred tax liabilities, projected future taxable income, tax planning
strategies and recent financial performance. Forming a conclusion that a
valuation allowance is not required is difficult when there is negative evidence
such as cumulative losses in recent years. Because
74
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
of our history of
losses, the net deferred tax assets have been fully offset by a valuation
allowance. The valuation allowance increased by $18,900,000, $21,000,000 and
$22,900,000 during the years ended December 31, 2009, 2008 and 2007,
respectively. Approximately $5,500,000 of the valuation allowance for deferred
tax assets relates to benefits of stock option deductions which, when
recognized, will be allocated directly to contributed capital.
As of December 31, 2009, we had domestic federal net operating loss
carryforwards of approximately $467,800,000 expiring at various dates beginning
in 2010 through 2029, and state net operating loss carryforwards of
approximately $187,700,000 expiring at various dates beginning in 2012 through
2029, if not utilized. Our foreign net operating loss carryforwards of
approximately $41,300,000 carry forward indefinitely. We also had federal
research and development tax credit carryforwards of approximately $14,100,000
expiring at various dates beginning in 2010 through 2029, if not utilized. Our
state research and development tax credit carryforwards of approximately
$11,800,000 carry forward indefinitely.
Due to the change of ownership provisions of the Tax Reform Act of 1986,
utilization of a portion of our domestic net operating loss and tax credit
carryforwards may be limited in future periods. Further, a portion of the
carryforwards may expire before being applied to reduce future income tax
liabilities.
We do not currently expect any significant changes to unrecognized tax
benefits during the fiscal year ended December 31, 2010. In certain cases, our
uncertain tax positions are related to tax years that remain subject to
examination by the relevant tax authorities. Tax years for which we have
carryforward net operating loss and credit attributes remain subject to
examination by federal and most state tax authorities. In significant foreign
jurisdictions, primarily Scotland and Hong Kong, the 2003 through 2008 tax years
generally remain subject to examination by their respective tax
authorities.
11. SEGMENT INFORMATION
Our executive management team represents our chief decision maker. To
date, we have viewed our operations as one segment, the discovery and
development of therapeutic and diagnostic products for oncology and human
embryonic stem cell therapies. As a result, the financial information disclosed
herein materially represents all of the financial information related to our
principal operating segment.
12. CONSOLIDATED STATEMENTS OF CASH FLOWS
DATA
| Year Ended December 31, | |||||||||||
| 2009 | 2008 | 2007 | |||||||||
| (In thousands) | |||||||||||
| Supplemental operating activities: | |||||||||||
| Cash in transit | $ | — | $ | — | $ | 7 | |||||
| Issuance of common stock and warrants to purchase common stock | |||||||||||
| for services rendered to date or to be received in future periods | $ | 3,350 | $ | 7,854 | $ | 5,121 | |||||
| Unrealized gain (loss) on investments in licensees | $ | 27 | $ | (11 | ) | $ | (9 | ) | |||
| Reclassification between derivative liabilities and equity, net | $ | 130 | $ | — | $ | 21,974 | |||||
| Issuance of common stock for 401(k) contributions and year-end | |||||||||||
| bonuses | $ | 3,707 | $ | 3,137 | $ | 3,590 | |||||
| Reclassification of deposits to other current assets | $ | 496 | $ | — | $ | — | |||||
| Supplemental investing activities: | |||||||||||
| Net unrealized (loss) gain on available-for-sale securities | $ | (472 | ) | $ | 27 | $ | 244 | ||||
| Supplemental financing activities: | |||||||||||
| Deemed dividend on derivatives | $ | 190 | $ | — | $ | 9,081 | |||||
There was no interest expense for the years ended December 31, 2009, 2008
and 2007.
75
GERON CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
13. SELECTED QUARTERLY FINANCIAL INFORMATION
(UNAUDITED)
| First | Second | Third | Fourth | ||||||||||||
| Quarter | Quarter | Quarter | Quarter | ||||||||||||
| (In thousands, except per share amounts) | |||||||||||||||
| Year Ended December 31, 2009 | |||||||||||||||
| Revenues | $ | 444 | $ | 183 | $ | 494 | $ | 605 | |||||||
| Operating expenses | 17,149 | 18,940 | 16,894 | 18,977 | |||||||||||
| Net loss | (16,811 | ) | (19,758 | ) | (15,224 | ) | (18,391 | ) | |||||||
| Deemed dividend on derivatives | — | (190 | ) | — | — | ||||||||||
| Net loss applicable to common stockholders | (16,811 | ) | (19,948 | ) | (15,224 | ) | (18,391 | ) | |||||||
| Basic and diluted net loss per share applicable to | |||||||||||||||
| common stockholders | $ | (0.20 | ) | $ | (0.23 | ) | $ | (0.17 | ) | $ | (0.20 | ) | |||
| Year Ended December 31, 2008 | |||||||||||||||
| Revenues | $ | 1,694 | $ | 198 | $ | 367 | $ | 544 | |||||||
| Operating expenses | 17,643 | 15,656 | 18,301 | 18,247 | |||||||||||
| Net loss applicable to common stockholders | (13,674 | ) | (13,564 | ) | (17,151 | ) | (17,632 | ) | |||||||
| Basic and diluted net loss per share applicable to | |||||||||||||||
| common stockholders | $ | (0.18 | ) | $ | (0.17 | ) | $ | (0.22 | ) | $ | (0.22 | ) | |||
Basic and diluted net losses per share are computed independently for
each of the quarters presented. Therefore, the sum of the quarters may not be
equal to the full year net loss per share amounts.
14. SUBSEQUENT EVENTS
We have evaluated events occurring from January 1, 2010 to February 26,
2010, the filing date of this Form 10-K, and have determined the following
events to be disclosed.
Vendor Stock Issuances
In January 2010, we issued 133,357 shares of our common stock to MPI
Research, Inc. (MPI) in a private placement as advance consideration under an
amendment to a master services agreement under which MPI has provided and will
continue to provide certain preclinical services in support of our clinical
programs. The total fair value of the common stock was $829,000 which has been
recorded as a prepaid asset and is being amortized to research and development
expense on a pro-rata basis as services are performed, which is expected to be
approximately six months.
In January 2010, we issued 94,741 shares of our common stock to Exponent,
Inc. (Exponent), the lessor of the premises at 149 Commonwealth Drive, as a
first installment payment of rent due under the extended lease agreement for the
period from May 1, 2010 through July 31, 2012. The fair value of the common
stock was $589,000 which has been recorded as a prepaid asset and will be
amortized to rent expense on a pro-rata basis over the lease term.
In January 2010, we issued 287,401 shares of our common stock to
Samchully Pharm Co. Ltd. (Samchully) in a private placement as advance
consideration related to an addendum agreement to a manufacturing agreement
pursuant to which Samchully is performing certain services and manufacturing
certain raw materials and products for us intended for use in human clinical
trials. The total fair value of the common stock was $1,704,000 which has been
recorded as a prepaid asset and is being amortized to research and development
expense on a pro-rata basis upon the performance of services and the proper
receipt of materials, which is expected to be over nine months.
Warrant Exchange and Direct Equity
Issuance
On January 14, 2010, we exchanged outstanding warrants to purchase
5,559,426 shares of common stock held by certain institutional investors for
2,700,000 shares of common stock. In connection with the exchange, we sold an
additional 1,481,481 shares of common stock to the investors at a premium to the
market price for gross proceeds of $10,000,000, and issued warrants to the
investors to purchase an additional 740,741 shares of common stock. The warrants
are immediately exercisable, expire on October 31, 2010 and have an exercise
price of $6.75 per share of common stock. A number of shares of common stock
equal to those issued in exchange for the outstanding warrants cannot be sold
during the 12 month period from the date of issuance unless the sales are at
prices in excess of $9.11 per share.
76
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
Not Applicable.
ITEM 9A. CONTROLS AND PROCEDURES
(I) Evaluation of Disclosure Controls and
Procedures
We maintain disclosure controls and procedures to ensure that information
we are required to disclose in reports that we file or submit under the
Securities Exchange Act of 1934, as amended, (Exchange Act) is recorded,
processed, summarized and reported within the time periods specified in
Securities and Exchange Commission’s (SEC) rules and forms. Our management
evaluated, with the participation of our chief executive officer (CEO) and our
chief financial officer (CFO), the effectiveness of our disclosure controls and
procedures, as such term is defined under Rule 13a-15(e) under the Exchange Act.
Based on that evaluation, our CEO and CFO concluded that our disclosure controls
and procedures were effective, at a reasonable assurance level, as of December
31, 2009 and as of the date of this filing.
There have been no significant changes in Geron’s internal control over
financial reporting that have materially affected, or are reasonably likely to
materially affect internal control over financial reporting during the fiscal
quarter ended December 31, 2009.
(II) Management’s Report on Internal Control
over Financial Reporting
Internal control over financial reporting refers to the process designed
by, or under the supervision of, our CEO and CFO, and effected by our Board of
Directors, management and other personnel, to provide reasonable assurance
regarding the reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with generally accepted
accounting principles, and includes those policies and procedures
that:
| (1) | Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the Company; | ||
| (2) | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and | ||
| (3) | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements. | ||
Management is responsible for establishing and maintaining an adequate
internal control over financial reporting for the Company. Internal control over
financial reporting cannot provide absolute assurance of achieving financial
reporting objectives because of its inherent limitations. Internal control over
financial reporting is a process that involves human diligence and compliance
and is subject to lapses in judgment and breakdowns resulting from human
failures. Internal control over financial reporting also can be circumvented by
collusion or improper management override. Because of such limitations, there is
a risk that material misstatements may not be prevented or detected on a timely
basis by internal control over financial reporting. However, these inherent
limitations are known features of the financial reporting process. Therefore, it
is possible to design into the process safeguards to reduce, though not
eliminate, this risk.
Under the supervision and with the participation of our management,
including our principal executive officer and principal financial officer, we
conducted an evaluation of the effectiveness of our internal control over
financial reporting based on the framework set forth in “Internal Control —
Integrated Framework” issued by the Committee of Sponsoring Organizations of the
Treadway Commission. Based on our evaluation
77
under the framework set
forth in “Internal Control — Integrated Framework,” our management concluded
that our internal control over financial reporting was effective as of December
31, 2009. The effectiveness of our internal control over financial reporting as
of December 31, 2009 has been audited by Ernst & Young LLP, an independent
registered public accounting firm, as stated in their report which is included
herein.
| THOMAS B. OKARMA | DAVID L. GREENWOOD |
| President and Chief Executive Officer | Executive Vice President |
| Chief Financial Officer |
(III) Report of Independent Registered Public
Accounting Firm
The Board of Directors
and Stockholders of Geron Corporation
We have audited Geron Corporation’s internal control over financial
reporting as of December 31, 2009, based on criteria established in Internal
Control—Integrated Framework issued by the Committee of Sponsoring Organizations
of the Treadway Commission (the COSO criteria). Geron Corporation’s management
is responsible for maintaining effective internal control over financial
reporting, and for its assessment of the effectiveness of internal control over
financial reporting included in the accompanying Management’s Report on Internal
Control Over Financial Reporting. Our responsibility is to express an opinion on
the company’s internal control over financial reporting based on our
audit.
We conducted our audit in accordance with the standards of the Public
Company Accounting Oversight Board (United States). Those standards require that
we plan and perform the audit to obtain reasonable assurance about whether
effective internal control over financial reporting was maintained in all
material respects. Our audit included obtaining an understanding of internal
control over financial reporting, assessing the risk that a material weakness
exists, testing and evaluating the design and operating effectiveness of
internal control based on the assessed risk, and performing such other
procedures as we considered necessary in the circumstances. We believe that our
audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process
designed to provide reasonable assurance regarding the reliability of financial
reporting and the preparation of financial statements for external purposes in
accordance with generally accepted accounting principles. A company’s internal
control over financial reporting includes those policies and procedures that (1)
pertain to the maintenance of records that, in reasonable detail, accurately and
fairly reflect the transactions and dispositions of the assets of the company;
(2) provide reasonable assurance that transactions are recorded as necessary to
permit preparation of financial statements in accordance with generally accepted
accounting principles, and that receipts and expenditures of the company are
being made only in accordance with authorizations of management and directors of
the company; and (3) provide reasonable assurance regarding prevention or timely
detection of unauthorized acquisition, use, or disposition of the company’s
assets that could have a material effect on the financial
statements.
Because of its inherent limitations, internal control over financial
reporting may not prevent or detect misstatements. Also, projections of any
evaluation of effectiveness to future periods are subject to the risk that
controls may become inadequate because of changes in conditions, or that the
degree of compliance with the policies or procedures may
deteriorate.
In our opinion, Geron Corporation maintained, in all material respects,
effective internal control over financial reporting as of December 31, 2009,
based on the COSO criteria.
We also have audited, in accordance with the standards of the Public
Company Accounting Oversight Board (United States), the consolidated balance
sheets of Geron Corporation as of December 31, 2009 and 2008, and the related
consolidated statements of operations, stockholders’ equity, and cash flows for
each of the three years in the period ended December 31, 2009 of Geron
Corporation and our report dated February 26, 2010 expressed an unqualified
opinion thereon.
| /s/ Ernst & Young LLP | |
| Palo Alto, California | |
| February 26, 2010 | |
78
ITEM 9B. OTHER INFORMATION
Not Applicable.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND
CORPORATE GOVERNANCE
Identification of Directors
The information required by this Item concerning our directors is
incorporated by reference from the section captioned “Proposal 1: Election of
Directors” contained in our Definitive Proxy Statement related to the Annual
Meeting of Stockholders to be held May 19, 2010, to be filed with the Securities
and Exchange Commission (the Proxy Statement).
Identification of Executive
Officers
The information required by this Item
concerning our executive officers is set forth in Part I of this
Report.
Code of Ethics
We have adopted a Code of Conduct with which every person who works for
Geron is expected to comply. The Code of Conduct is publicly available on our
website under the Investor Relations section at www.geron.com. This website
address is intended to be an inactive, textual reference only; none of the
material on this website is part of this Report. If any substantive amendments
are made to the Code of Conduct or any waiver granted, including any implicit
waiver, from a provision of the Code to our Chief Executive Officer, Chief
Financial Officer or Corporate Controller, we will disclose the nature of such
amendment or waiver on that website or in a report on Form 8-K.
Copies of the Code of Conduct will be furnished without charge to any
person who submits a written request directed to the attention of our Secretary,
at our offices located at 230 Constitution Drive, Menlo Park, California,
94025.
Section 16(a) Compliance
Information concerning Section 16(a) beneficial ownership reporting
compliance is incorporated by reference from the section captioned “Section
16(a) Beneficial Ownership Reporting Compliance” contained in the Proxy
Statement.
Audit Committee Report
The information required by this Item is incorporated by reference from
the section captioned “Audit Committee Report” contained in the Proxy
Statement.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this Item is incorporated by reference from
the sections captioned “Certain Transactions,” “Compensation Discussion and
Analysis,” “Executive Compensation” and “Compensation Committee Report”
contained in the Proxy Statement.
| ITEM 12. |
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL
OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER
MATTERS
|
The information required by this Item is incorporated by reference from
the sections captioned “Security Ownership of Certain Beneficial Owners and
Management” and “Equity Compensation Plans” contained in the Proxy
Statement.
79
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED
TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this Item is incorporated by reference from
the sections captioned “Proposal 1: Election of Directors,” “Certain
Transactions” and “Executive Compensation” contained in the Proxy Statement.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND
SERVICES
The information required by this Item is incorporated by reference from
the section captioned “Principal Accountant Fees and Services” contained in the
Proxy Statement.
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT
SCHEDULES
(a) (1) Consolidated Financial
Statements
Included in Part II, Item 8 of this
Report:
| Page | |
| Report of Independent Registered Public Accounting Firm | 50 |
| Consolidated Balance Sheets — December 31, 2009 and 2008 | 51 |
| Consolidated Statements of Operations — Years ended December 31, 2009, 2008 and 2007 | 52 |
| Consolidated Statements of Stockholders’ Equity — Years ended December 31, 2009, 2008 and 2007 | 53 |
| Consolidated Statements of Cash Flows — Years ended December 31, 2009, 2008 and 2007 | 54 |
| Notes to Consolidated Financial Statements | 55 |
(2) Financial Statement
Schedules
Financial statement schedules are omitted because they are not required
or the information is disclosed in the financial statements listed in Item
15(a)(1) above.
(3) Exhibits
See Exhibit Index.
(b) Reports on Form 8-K
None.
(c) Index to Exhibits
See Exhibits listed under Item
15(a)(3) above.
(d) Financial Statements and
Schedules
The financial statement schedules
required by this Item are listed under Item 15(a)(1) and (2) above.
80
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities
Exchange Act of 1934, the Registrant has duly caused this report to be signed on
its behalf by the undersigned, thereunto duly authorized.
| GERON CORPORATION | |||
| Date: February 26, 2010 | By: | /S/ THOMAS B. OKARMA | |
| THOMAS B. OKARMA | |||
| President and Chief Executive Officer | |||
POWER OF ATTORNEY
KNOW BY ALL PERSONS BY THESE PRESENTS, that each person whose signature
appears below constitutes and appoints, jointly and severally, Thomas B. Okarma
and David L. Greenwood, and each one of them, attorneys-in-fact for the
undersigned, each with the power of substitution, for the undersigned in any and
all capacities, to sign any and all amendments to this annual report on Form
10-K, and to file the same, with exhibits thereto and other documents in
connection therewith, with the Securities and Exchange Commission, hereby
ratifying and confirming all that each of said attorneys-in-fact, or his
substitutes, may do or cause to be done by virtue hereof.
IN WITNESS WHEREOF, each of the undersigned has executed this Power of
Attorney as of the date indicated opposite his/her name.
Pursuant to the requirements of the Securities Exchange Act of 1934, this
report has been signed below by the following persons on behalf of the
Registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /S/ THOMAS B. OKARMA | President, Chief Executive Officer and Director | February 26, 2010 | ||
| THOMAS B. OKARMA | (Principal Executive Officer) | |||
| /S/ DAVID L. GREENWOOD | Executive Vice President, Chief Financial Officer, | February 26, 2010 | ||
| DAVID L. GREENWOOD | Treasurer and Secretary | |||
| (Principal Financial and Accounting Officer) | ||||
| /S/ ALEXANDER E. BARKAS | Director | February 26, 2010 | ||
| ALEXANDER E. BARKAS | ||||
| /S/ KARIN EASTHAM | Director | February 26, 2010 | ||
| KARIN EASTHAM | ||||
| /S/ EDWARD V. FRITZKY | Director | February 26, 2010 | ||
| EDWARD V. FRITZKY | ||||
| /S/ CHARLES J. HOMCY | Director | February 26, 2010 | ||
| CHARLES J. HOMCY | ||||
| /S/ THOMAS D. KILEY | Director | February 26, 2010 | ||
| THOMAS D. KILEY | ||||
| /S/ PATRICK J. ZENNER | Director | February 26, 2010 | ||
| PATRICK J. ZENNER | ||||
81
EXHIBIT INDEX
| Incorporation by Reference | ||||||||
| Exhibit | Exhibit | |||||||
| Number | Description | Number | Filing | Filing Date | ||||
| 3.1 | Amended and Restated Certificate of Incorporation of the Registrant | 3.1 | S-1 | June 12, 1996 | ||||
| 3.2 | Certificate of Amendment of Restated Certificate of Incorporation of the Registrant | 3.1 | 10-Q | July 31, 2006 | ||||
| 3.3 | Bylaws of Registrant | 3.3 | 10-K | March 13, 2000 | ||||
| 4.1 | Form of Common Stock Certificate | 4.1 | S-1 | June 12, 1996 | ||||
| 4.2 | Rights Agreement, dated as of July 20, 2001, by and between the Registrant and U.S. Stock Transfer Corporation, as Rights Agent, which includes the form of Certification of Designations of the Series A Junior Participating Preferred Stock of the Registrant as Exhibit A, the form of Right Certificate as Exhibit B and the Summary of Rights to Purchase Preferred Shares as Exhibit C | 4.1 | 8-K | July 23, 2001 | ||||
| 4.3 | Form of Senior Indenture, between the Registrant and one or more trustees to be named | 4.5 | S-3 | July 9, 2009 | ||||
| 4.4 | Form of Subordinated Indenture, between the Registrant and one or more trustees to be named | 4.6 | S-3 | July 9, 2009 | ||||
| 4.5 | Amended and Restated Warrant to purchase 100,000 shares of common stock issued by Registrant to private investor, Eve M. Patton dated April 13, 2009 | 4.1 | 10-Q | July 31, 2009 | ||||
| 4.6 | Amended and Restated Warrant to purchase 200,000 shares of common stock issued by Registrant to private investor, Eve M. Patton dated April 13, 2009 | 4.2 | 10-Q | July 31, 2009 | ||||
| 4.7 | Common Stock Warrant Agreement issued by the Registrant to University Technology Corporation, dated as of August 30, 2001 | 4.3 | S-3 | September 27, 2001 | ||||
| 4.8 | Form of Warrant, issued by the Registrant to certain Purchasers, dated April 22, 2005 | 4.2 | 8-K | April 25, 2005 | ||||
| 4.9 | Form of A Warrant, Amended and Restated, dated December 21, 2007 issued by the Registrant to certain Purchasers | 4.12 | 10-K | February 28, 2008 | ||||
| 4.10 | Form of A Warrant, Amended and Restated, dated December 21, 2007 issued by the Registrant to certain Purchasers | 4.13 | 10-K | February 28, 2008 | ||||
| 4.11 | Form of D Warrant, Amended and Restated, dated December 21, 2007 issued by the Registrant to certain Purchasers | 4.14 | 10-K | February 28, 2008 | ||||
| 4.12 | Form of Common Stock Purchase Warrant issued by Registrant to certain Purchasers, dated September 9, 2009 | 4.2 | 8-K | September 10, 2009 | ||||
| 4.13 | Form of Lock-Up Agreement issued by Registrant to certain Investors, dated September 9, 2009 | 4.3 | 8-K | September 10, 2009 | ||||
| 10.1 | Form of Indemnification Agreement | 10.1 | S-1 | June 12, 1996 | ||||
| 10.2 | 1992 Stock Option Plan, as amended | Appendix A | Def 14A | April 9, 2001 | ||||
| 10.3 | Amended and Restated 1996 Employee Stock Purchase Plan | 10.2 | 10-Q | July 31, 2009 | ||||
| 10.4 | 1996 Directors’ Stock Option Plan, as amended | Appendix B | Def 14A | April 15, 2003 | ||||
| 10.5 | Amended and Restated 2002 Equity Incentive Plan | 10.1 | 10-Q | April 30, 2007 | ||||
| 10.6 | Amended and Restated 2006 Directors’ Stock Option Plan | 10.1 | 10-Q | July 31, 2009 | ||||
| 10.7† | Patent License Agreement dated September 8, 1992 between the Registrant and University of Texas Southwestern Medical Center at Dallas | 10.7 | S-1 | June 12, 1996 | ||||
82
| 10.8† | Intellectual Property License Agreement dated December 9, 1996 between the Registrant and University Technology Corporation | 10.30 | 10-Q | May 13, 1997 | ||||
| 10.9† | Exclusive License Agreement dated February 2, 1994 between the Registrant and the Regents of the University of California | 10.9 | S-1 | June 12, 1996 | ||||
| 10.10† | License Agreement dated August 1, 1997 between the Registrant and The Johns Hopkins University | 10.35 | 10-Q | November 14, 1997 | ||||
| 10.11† | License Agreement dated May 3, 1999, among the Registrant, Roslin Bio-Med Ltd. and the Roslin Institute | 10.43 | 8-K | May 18, 1999 | ||||
| 10.12† | First Amendment to Intellectual Property License Agreement dated July 23, 2001, by and among the Registrant and University Technology Corporation | 4.1 | S-3 | September 27, 2001 | ||||
| 10.13† | License Agreement dated as of January 8, 2002, by and between the Registrant and Wisconsin Alumni Research Foundation | 10.1 | 8-K | January 18, 2002 | ||||
| 10.14† | License Amendment Agreement between the Registrant and Transgenomic, Inc., dated June 2, 2003 | 10.1 | 10-Q | July 30, 2003 | ||||
| 10.15† | License Agreement dated as of March 6, 2004 by and between the Registrant and Merix Bioscience, Inc. | 10.4 | 10-Q | July 30, 2004 | ||||
| 10.16† | Research, Development and Commercialization License Agreement dated July 15, 2005 between the Registrant and Merck & Co., Inc. | 10.1 | 10-Q | August 5, 2005 | ||||
| 10.17 | Restructuring Agreement between Biotechnology Research Corporation and Registrant, dated June 15, 2007 | 10.1 | 10-Q | July 31, 2007 | ||||
| 10.18 | Amended and Restated Joint Venture Agreement between Biotechnology Research Corporation, Registrant and TA Therapeutics, Ltd., dated June 15, 2007 | 10.2 | 10-Q | July 31, 2007 | ||||
| 10.19 | Contribution Agreement between Registrant and ViaGen, Inc. dated August 8, 2008 | 10.1 | 8-K | August 12, 2008 | ||||
| 10.20† | Exclusive License and Alliance Agreement by and between Registrant and GE Healthcare UK Limited, dated June 29, 2009 | 10.1 | 8-K | July 2, 2009 | ||||
| 10.21 | Series A Preferred Stock Purchase Agreement by and between ViaGen, Inc. and Registrant, dated September 16, 2009 | 10.1 | 10-Q | October 30, 2009 | ||||
| 10.22 | Employment agreement between Registrant and Thomas Okarma, dated January 21, 2003 | 10.1 | 10-Q | April 30, 2003 | ||||
| 10.23 | Employment agreement between Registrant and David Greenwood, dated January 21, 2003 | 10.2 | 10-Q | April 30, 2003 | ||||
| 10.24 | Employment agreement between Registrant and David Earp, dated January 21, 2003 | 10.3 | 10-Q | April 30, 2003 | ||||
| 10.25 | Employment agreement between Registrant and Melissa Kelly, dated January 21, 2003 | 10.5 | 10-Q | April 30, 2003 | ||||
| 10.26 | Employment agreement between Registrant and Jane Lebkowski, dated January 21, 2003 | 10.6 | 10-Q | April 30, 2003 | ||||
| 10.27 | Amendment to employment agreement between Registrant and Thomas Okarma, dated December 19, 2008 | 10.21 | 10-K | February 27, 2009 | ||||
| 10.28 | Amendment to employment agreement between Registrant and David Greenwood, dated December 19, 2008 | 10.22 | 10-K | February 27, 2009 | ||||
| 10.29 | Amendment to employment agreement between Registrant and David Earp, dated December 19, 2008 | 10.23 | 10-K | February 27, 2009 |
83
| 10.30 | Amendment to employment agreement between Registrant and Melissa Kelly Behrs, dated December 19, 2008 | 10.25 | 10-K | February 27, 2009 | ||||
| 10.31 | Amendment to employment agreement between Registrant and Jane Lebkowski, dated December 19, 2008 | 10.26 | 10-K | February 27, 2009 | ||||
| 10.32 | Offer letter agreement between Registrant and Stephen Kelsey, dated April 8, 2009 | 10.3 | 10-Q | July 31, 2009 | ||||
| 10.33 | Amended and Restated Severance Plan, effective December 19, 2008 | 10.27 | 10-K | February 27, 2009 | ||||
| 10.34 | Fifth Amendment to Lease by and between the Registrant and David D. Bohannon Organization, dated March 19, 2008 | 10.1 | 10-Q | April 30, 2008 | ||||
| 10.35 | Second Amendment to Lease by and between the Registrant and David D. Bohannon Organization, dated March 19, 2008 | 10.2 | 10-Q | April 30, 2008 | ||||
| 14.1 | Code of Conduct | 14.1 | 10-K | February 27, 2004 | ||||
| 21.1 | List of Subsidiaries | 21.1 | 10-K | February 28, 2008 | ||||
| 23.1 | Consent of Independent Registered Public Accounting Firm | |||||||
| 24.1 | Power of Attorney (see signature page) | |||||||
| 31.1 | Certification of Chief Executive Officer pursuant to Form of Rule 13a-14(a), as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002, dated February 26, 2010 | |||||||
| 31.2 | Certification of Chief Financial Officer pursuant to Form of Rule 13a-14(a), as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002, dated February 26, 2010 | |||||||
| 32.1 | Certification of Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, dated February 26, 2010 | |||||||
| 32.2 | Certification of Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, dated February 26, 2010 |
| † | Certain portions of this Exhibit have been omitted for which confidential treatment has been requested and filed separately with the Securities and Exchange Commission. |
84