Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For Fiscal Year Ended December 31, 2009
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
From the transition period from to
Commission File Number 000-31255
ISTA PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 33-0511729 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
15295 Alton Parkway, Irvine, California 92618
(Address of principal executive offices)
(949) 788-6000
(Registrant’s telephone number)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common Stock, $0.001 par value | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company: in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ¨ | Accelerated filer | x | |||
| Non-accelerated filer | ¨ | Smaller reporting company | ¨ | |||
Indicate by check mark whether the registrant is a shell company (as defined by Rule 12b-2 of the Act). Yes ¨ No x
As of June 30, 2009, the aggregate market value of the Registrant’s voting stock held by non-affiliates was approximately $65,320,382.
As of January 29, 2010 there were 33,300,541 shares of Common Stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
None.
Table of Contents
| PART I | ||||
| Item 1: |
1 | |||
| Item 1A: |
11 | |||
| Item 1B: |
23 | |||
| Item 2: |
23 | |||
| Item 3: |
23 | |||
| Item 4: |
23 | |||
| PART II | ||||
| Item 5: |
25 | |||
| Item 6: |
28 | |||
| Item 7: |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
29 | ||
| Item 7A: |
45 | |||
| Item 8: |
45 | |||
| Item 9: |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
46 | ||
| Item 9A: |
46 | |||
| Item 9B: |
49 | |||
| PART III | ||||
| Item 10: |
49 | |||
| Item 11: |
49 | |||
| Item 12: |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
49 | ||
| Item 13: |
Certain Relationships and Related Transactions, and Director Independence |
49 | ||
| Item 14. |
49 | |||
| PART IV | ||||
| Item 15: |
50 | |||
Table of Contents
ISTA PHARMACEUTICALS, INC.
PART I
References in this Annual Report on Form 10-K to “ISTA”, “we”, “our”, “us”, or the “Company” refer to ISTA Pharmaceuticals, Inc. This Annual Report on Form 10-K contains forward-looking statements based on expectations, estimates and projections as of the date of this filing. Actual results may differ materially from those expressed in forward-looking statements. See Item 7 of Part II – “Management’s Discussion and Analysis of Financial Condition and Results of Operations – Forward-Looking Statements.” Xibrom (bromfenac sodium ophthalmic solution)®, Xibrom™, XiDay™, Istalol® , Vitrase®, Bepreve™, T-Pred™, ISTA®, ISTA Pharmaceuticals, Inc.® and the ISTA logo are our trademarks, either owned or under license.
We obtained the market data and industry information contained in this Annual Report on Form 10-K from internal surveys, estimates, reports and studies, as appropriate, as well as from market research, publicly available information and industry publications. Although we believe our internal surveys, estimates, reports, studies and market research, as well as industry publications are reliable, we have not independently verified such information, and as such, we do not make any representation as to its accuracy.
| Item 1: | Business. |
Overview
We are a commercial stage, multi-specialty pharmaceutical company developing, marketing and selling our own products in the United States. We manufacture our products through third party contracts, and we in-license or acquire new technology to add to our internal development efforts from time to time.
Our products and product candidates seek to treat serious diseases of the eye and allergies, and include therapies for ocular inflammation and pain, glaucoma, dry eye and ocular and nasal allergies. The U.S. prescription markets our therapies seek to address include key segments of the $5.5 billion ophthalmic pharmaceutical market and the $2.2 billion nasal allergy market.
We currently have four products available for sale in the United States and Puerto Rico: Xibrom (bromfenac sodium ophthalmic solution) for the treatment of inflammation and pain following cataract surgery, Bepreve (bepotastine besilate ophthalmic solution) for the treatment of ocular itching associated with allergic conjunctivitis, Istalol (timolol maleate ophthalmic solution) for the treatment of glaucoma, and Vitrase (hyaluronidase for injection) for use as a spreading agent.
We have incurred losses since inception and had an accumulated deficit of $397.3 million (including a non-cash valuation warrant adjustment of $52.1 million) through December 31, 2009.
Our Products and Pipeline
The following is a summary of our key products, late-stage clinical products and product candidates:
| Product/Product Candidate |
Indication |
Development Status | ||
| Xibrom (Twice-Daily) |
Ocular inflammation and pain following cataract surgery | Marketed | ||
| Bepreve |
Ocular itching associated with allergic conjunctivitis | Marketed | ||
| Istalol |
Glaucoma | Marketed | ||
| Vitrase |
Spreading agent | Marketed | ||
| XiDay (bromfenac once-daily) |
Ocular inflammation and pain following cataract surgery | Supplemental New Drug Application, or sNDA, filed with the U.S. Food and Drug Administration, or FDA | ||
| T-Pred |
Steroid-responsive inflammatory ocular conditions |
Phase III | ||
| Bromfenac lower concentration |
Dry eye syndrome | Announced positive, preliminary results from Phase II study | ||
| Ecabet Sodium |
Dry eye syndrome | Announced positive, preliminary results from Phase IIb confirmatory study | ||
| Bepotastine Nasal |
Allergic rhinitis | Nasal formulation in development | ||
1
Table of Contents
Xibrom (bromfenac sodium ophthalmic solution) 0.09% – twice-daily
Xibrom is a twice-daily topical non-steroidal anti-inflammatory formulation of bromfenac for the treatment of ocular inflammation and pain following cataract surgery. We received approval from the FDA for Xibrom for the treatment of ocular inflammation following cataract surgery in March 2005. We launched Xibrom in the United States in the second quarter of 2005. In January 2006, we received FDA approval of an expanded indication of Xibrom to include the treatment of pain following cataract surgery. We promote Xibrom through our own sales force.
Xibrom was first developed by Senju Pharmaceuticals, Co. Ltd., or Senju, and launched in Japan in 2000. In May 2002, we acquired marketing rights for Xibrom in the United States under a license agreement with Senju. In December 2009, we expanded the marketing rights to include not only the United States and its possessions, but also Canada and Mexico.
Based upon 2009 data from IMS Health, we estimate that 2009 sales in the U.S. topical ophthalmic non-steroidal anti-inflammatory market were approximately $326 million, with total prescriptions of 2.7 million. From 2008 to 2009, the U.S. topical ophthalmic non-steroidal anti-inflammatory market grew approximately 28% in dollars and 7% in total prescriptions. Certain other non-steroid treatments currently available must be dosed two, three or four times a day as compared to Xibrom’s twice-daily dosing.
In January 2009, the patent on Xibrom expired, exposing us to potential future generic competition. Most companies that manufacture drug substances file a Drug Master File, or DMF, with the FDA. These DMFs contain information on the manufacture and quality control of those drug substances. Most companies that file abbreviated New Drug Applications, or ANDAs, to gain approval to market a generic version of a previously patented pharmaceutical product will reference a DMF for the drug substance contained in the product. In July 2009, a new DMF was filed for bromfenac, the drug substance in Xibrom. Based upon publicly available information on the average FDA review times for ANDAs, we do not anticipate a generic form of bromfenac to gain approval by a competitor until 2011, or later.
For the year ended December 31, 2009, sales of Xibrom accounted for 73% of our total net revenues. We believe that sales of Xibrom will continue to be a significant portion of our total net revenues in 2010.
In December 2009, we announced that we submitted an sNDA to the FDA for bromfenac ophthalmic solution dosed once-daily as a treatment for ocular inflammation and pain following cataract surgery. See “XiDay (bromfenac sodium ophthalmic solution) 0.09% - once-daily” below for further information.
Bepreve (bepotastine besilate ophthalmic solution) 1.5%
Bepreve is a twice-daily prescription treatment for ocular itching associated with allergic conjunctivitis in patients two years of age and older. In September 2009, we received approval from the FDA for, and launched, Bepreve in the United States. We promote Bepreve through our own sales force to ophthalmologists, optometrists and allergists.
Bepreve was first approved in Japan for use as a systemic drug in the treatment of allergic rhinitis and urticaria/pruritus in July 2000 and January 2002, respectively, and is marketed by Mitsubishi Tanabe Pharma Corporation (formerly Tanabe Seiyaku Co., Ltd.) under the brand name TALION®. TALION was co-developed by Tanabe Seiyaku and Ube Industries, Ltd., who discovered the utility of bepotastine. In 2001, Tanabe Seiyaku granted Senju exclusive worldwide rights, with the exception of certain Asian countries, to develop, manufacture and market bepotastine for ophthalmic use. In 2006, we licensed the exclusive North American rights from Senju to an eye drop formulation of bepotastine for the treatment of allergic conjunctivitis. In 2007, we licensed exclusive North American rights to nasal dosage forms of bepotastine from Tanabe Seiyaku and obtained a future right to negotiate for a North American license to oral dosage forms of bepotastine for allergy treatment.
Based upon 2009 data from IMS Health, we estimate that 2009 sales in the U.S. ocular allergy market were approximately $594 million, with total prescriptions of 6.6 million. From 2008 to 2009, the U.S. ocular allergy market grew approximately 6% in total dollars and remained flat in total prescriptions.
Istalol (timolol)
Istalol is our once-daily eye drop solution of timolol, a beta-blocking agent for the treatment of glaucoma. Istalol was developed by Senju in Japan. In May 2002, we acquired marketing rights for Istalol in the United States under a license agreement with Senju.
We received FDA approval to market Istalol in the United States in 2004 for the treatment of glaucoma. We promote Istalol through our own sales force.
2
Table of Contents
Glaucoma is a chronic disease that gradually reduces eyesight without warning and often without symptoms. If undetected and untreated, glaucoma can lead to irreversible eye damage and eventual blindness. According to the Glaucoma Research Foundation, glaucoma is the cause of an estimated 9-12% of all blindness cases and is the second leading cause of blindness in the United States. Currently, its causes are not well understood and there is no known cure.
According to the Glaucoma Research Foundation, four million people in the United States suffer from the disease, with 120,000 new cases documented annually. According to prescription data compiled by IMS Health for 2009, we estimate that the U.S. pharmaceutical market for the treatment of glaucoma exceeds $1.9 billion per year. Of this amount, the ophthalmic beta-blocker market was approximately $173 million in 2009, primarily at generic prices, with over 4.4 million prescriptions written in 2009. Timolol maleate, which is currently available from several manufacturers in either a twice-daily eye drop solution or once-daily gel formulation, is the leading beta-blocker to treat glaucoma in the United States. Istalol, given once-daily, has shown efficacy and safety comparable to timolol maleate solution, given twice-daily. Other than Istalol, the only available formulations of timolol maleate that have demonstrated efficacy with once-daily dosing are gels, which have been known to cause blurring of patients’ vision.
Vitrase (ovine hyaluronidase)
We launched Vitrase, our proprietary formulation of ovine hyaluronidase, for use as a spreading agent in 2005. The term hyaluronidase describes a group of naturally occurring enzymes that can digest certain forms of carbohydrate molecules called proteoglycans. Vitrase, when used as a spreading agent, is injected into connective tissue, where it modifies the permeability of such tissues and promotes diffusion of injected drugs, thus accelerating their absorption.
In May 2004, the FDA approved our NDA for Vitrase, in a lyophilized 6,200 USP units multi-purpose vial, for use as a spreading agent to facilitate the absorption and dispersion of other injected drugs. In October 2004, the FDA informed us that Vitrase for use as a spreading agent was entitled to five-year new chemical market exclusivity under the federal Food, Drug and Cosmetic Act. In September 2009, we announced the discontinuation of Vitrase, lyophilized 6,200 USP units multi-purpose vial.
In December 2004, the FDA approved our sNDA for Vitrase for use as a spreading agent at a concentration of 200 USP units/mL in sterile solution. We continue to sell our 200 USP units/mL vial of Vitrase through our own sales force.
XiDay (bromfenac sodium ophthalmic solution) 0.09% – once-daily
We are developing a once-daily version of Xibrom, or XiDay, for the treatment of ocular inflammation and pain following cataract surgery. In August 2009, we announced positive preliminary Phase III results from our XiDay Phase III confirmatory clinical study. XiDay achieved statistical significance in the study’s primary endpoint, the absence of ocular inflammation 15 days following cataract surgery, and the secondary efficacy endpoint, the elimination of ocular pain one day post surgery. In December 2009 we filed a sNDA with the FDA seeking approval of the XiDay formulation for once-daily treatment for the inflammation and pain following cataract surgery. We expect to be notified by the FDA by the end of February or early March 2010 about the Prescription Drug User Fee Act, or PDUFA, date.
T-Pred (tobramycin and prednisolone acetate combination product)
T-Pred is our proprietary formulation of a fixed combination product of tobramycin 0.3% and prednisolone acetate 1.0%. T-Pred is being developed for the treatment of steroid-responsive inflammatory ocular conditions where risk of bacterial infection exists.
T-Pred, if approved by the FDA, will compete in the antibiotic/steroid combination segment of the U.S. topical ophthalmic anti-inflammatory market. Based upon management estimates and 2009 prescription data compiled by IMS Health, we estimate that 2009 sales in the U.S. topical ophthalmic anti-inflammatory market were approximately $753 million, with total prescriptions of 10.8 million. In 2009, the combination antibiotic and steroid segment of the ophthalmic anti-inflammatory market had approximately a 37% share of the prescriptions or $272 million in prescription dollars and about 3.9 million prescriptions according to data compiled by IMS Health. Assuming approval by the FDA, we plan to promote this product with our own sales force.
In February 2006, we announced positive results from our Phase III bioequivalence study of T-Pred for the treatment of steroid-responsive inflammatory ocular conditions where risk of bacterial infection exists.
In July 2006, we submitted to the FDA an NDA for T-Pred for the treatment of steroid-responsive inflammatory ocular conditions where risk of bacterial infection exists.
3
Table of Contents
In May 2007, we received a not approvable letter from the FDA. The FDA assessed our clinical data and found that it did not show sufficient equivalence between the prednisolone component in T-Pred and PredForte at least at one of the time points measured. In addition, the FDA found that our clinical data did not show sufficient equivalence in the kill time between the tobramycin components in T-Pred and Tobrex®, although it did show equivalence versus Zylet® and Tobradex®. After further discussions with the FDA, we initiated an additional clinical study and in vitro work to address the issues raised by the FDA.
In September 2009 we announced the preliminary results from two completed studies. In the first study, we evaluated the antimicrobial equivalence between T-Pred and a tobramycin-containing reference product. We successfully demonstrated the antimicrobial bioequivalence of T-Pred to the reference product in each of the 26 required tests. The second study was a Phase 3 clinical study designed to determine the bioequivalence of prednisolone concentrations between T-Pred and a reference product containing prednisolone acetate 1.0%. Although T-Pred’s prednisolone concentrations in this study were similar to those of the reference product, bioequivalence was not demonstrated. We believe differences in the prednisolone reference product, which included a higher drug concentration in the formulation and a recent change in the commercial product delivery dose of the reference product, may have contributed to the variations in study results between the two products. We are discussing the study results with the FDA to determine the best path forward for T-Pred and determining the appropriate clinical study or studies to perform in lieu of an additional prednisolone bioequivalence study, although no further clinical studies are planned for 2010.
Bromfenac – lower concentration
We are developing a lower concentration of bromfenac for the treatment of dry eye syndrome. According to the National Eye Institute, dry eye syndrome, which is also referred to as keratoconjunctivities sicca, or KCS, is defined as a disorder of the tear film due to the tear deficiency or excessive tear evaporation which causes damage to the interpalpebral, or the exposed area between the upper and lower eye lids, ocular surface and is associated with symptoms of ocular discomfort. Dry eye syndrome has been linked with a number of factors, including age, hormonal changes, ocular disease, medications that disrupt tear secretion or blinking, and autoimmune diseases such as lupus and rheumatoid arthritis. In severe cases of dry eye syndrome, scarring develops that may lead to blindness. Based on data compiled from various publicly available sources, we estimate that annual sales in the U.S. prescription dry eye market were approximately $502 million in 2009, with total prescriptions of over 2.5 million.
In June 2009, we announced positive results from a proof-of-concept Phase II clinical study in subjects with dry eye disease. The study achieved statistical significance in the primary endpoint of the objective sign of conjunctival staining as compared to baseline. The study also achieved statistical significance on the objective sign of corneal staining as compared to baseline. Patients also achieved statistically significant improvements in subjective symptoms measured by the Ocular Surface Disease Index and improvements in patients’ most bothersome ocular symptoms.
We plan to initiate Phase III studies with bromfenac for the treatment of dry eye syndrome, which could start in the first half of 2010.
Ecabet sodium
We are developing ecabet sodium as a prescription eye drop for the treatment of dry eye syndrome. Ecabet sodium represents a new class of molecules that increases the quantity and quality of mucin produced by conjunctival goblet cells and corneal epithelia. Mucin is a glycoprotein component of tear film that lubricates while retarding moisture loss from tear evaporation. Ecabet sodium is currently marketed in Japan as an oral agent for treatment of gastric ulcers and gastritis. In November 2004, we acquired U.S. marketing rights to ecabet sodium for the treatment of dry eye syndrome in the United States under a license agreement with Senju.
In February 2006, we announced positive preliminary results from our U.S. randomized, three-arm (placebo/3.0%/3.9%) Phase IIb clinical study of ecabet sodium for the treatment of dry eye syndrome. The preliminary results of our Phase IIb study demonstrated a strong trend in efficacy for the lower (3%) dose with respect to ecabet sodium’s ability to address two objective signs of dry eye syndrome: corneal staining and blink rate. In addition, patients treated with the lower dose of ecabet sodium reported positive trends for reductions in symptoms as measured by an objective assessment using the Ocular Symptom Disease Index, or OSDI, and a subjective assessment of patients’ most bothersome symptom. In the preliminary results, no efficacy trends versus placebo were observed with respect to the higher (3.9%) dose. In addition, preliminary findings from our Phase IIb study suggested a favorable safety profile for ecabet sodium for the treatment of dry eye syndrome.
During the third quarter of 2006, we initiated a 100 patient Phase IIb confirmatory study designed to finalize the entry criteria and other clinical parameters for future registration studies. In May 2007, we announced positive results from the preliminary analysis of our Phase IIb confirmatory study of ecabet sodium. Patients in the ecabet sodium group achieved a
4
Table of Contents
strong trend in the objective sign of blink rate. In addition, patients in the ecabet sodium group reported a strong trend in the OSDI and a positive trend in the subjective assessment of patients’ most bothersome symptom. Strong and positive trends are used to confirm observations from previous clinical ecabet sodium studies and to serve as indicators of potential efficacy endpoints in Phase III studies. While our Phase IIb confirmatory study was not powered to show statistical significance, ecabet sodium did achieve statistical significance in the OSDI assessment. There were no reports of serious ocular adverse events compared with the placebo.
In January 2009, we announced positive results from our Phase II study. Patients treated with ecabet sodium achieved a strong positive trend in the objective sign of Tear Film Break-Up Time and a positive trend in the objective sign of the Schirmer Test, which measures the quantity of tears produced. In contrast, there were no trends seen in the placebo group for either objective sign. In addition, there were no trends seen in either group in subjective symptoms as measured by the OSDI or patient’s most bothersome reported symptom. In Phase II tests where observations are not powered to show statistical significance, strong and positive trends are used as indicators of potential efficacy to consider conducting subsequent Phase III studies.
After reviewing the guidance from, and our discussions with, the FDA, we believe that by conducting two successful Phase III environmental clinical studies for improvement in signs, and two successful Phase III controlled chamber clinical studies for the improvement in symptoms, we could receive marketing approval. We are continuing our work on the design of future Phase III studies with ecabet sodium.
Bepotastine nasal
In addition to our ophthalmic solution development program, we are developing a proprietary nasal formulation of bepotastine for the treatment of allergic rhinitis. In September 2007, we obtained exclusive North American rights to nasal dosage forms of bepotastine, an investigational product for the treatment of allergy symptoms, from Mitsubishi Tanabe Pharma Corporation (formerly Tanabe Seiyaku Co., Ltd.). Based upon 2009 data from IMS, we estimate the U.S. allergic rhinitis market to be approximately $2.2 billion in sales, with the nasal antihistamine component comprising about 12% of all prescriptions.
Bepotastine nasal is currently in formulation development, with animal studies planned in 2010. We also plan to initiate human clinical studies during 2010.
Other Product Candidates and Development Activities
In addition to the products presently in human clinical trials, we have a number of products that may be ready for late stage clinical study initiation in the future. These include a strong steroid product candidate to treat ocular inflammation, iganidipine, to enhance ocular nerve blood flow, and a new formulation of latanoprost, a prostaglandin, for the treatment of glaucoma.
We continually evaluate opportunities for late-stage or currently-marketed complementary products and for expansion of our existing ophthalmology, optometry, and allergy product franchises. We plan to continue to pursue such opportunities through further licensing arrangements, collaborations and product acquisitions, along with related development activities. Our ability to execute on such opportunities in some circumstances may be dependent upon our ability to raise additional capital on commercially reasonable terms.
Product Licensing Agreements
Xibrom, Istalol, Bepreve, Ecabet Sodium, Prostaglandins and Iganidipine Agreements With Senju
In May 2002, we acquired substantially all of the assets of AcSentient, Inc., or AcSentient, which included exclusive U.S. development, manufacturing and marketing rights for Istalol and Xibrom. Istalol and Xibrom were originally licensed by AcSentient from Senju. The full rights and obligations of AcSentient under the Senju license agreements were assigned to us as a part of the acquisition agreement between us and AcSentient, with such transfer approved by Senju.
In November 2004, we entered into another license agreement with Senju under which Senju granted to us exclusive U.S. marketing rights to Senju’s ecabet sodium product for the treatment of dry eye syndrome.
In 2006, we entered into three additional license agreements with Senju under which Senju has granted us exclusive North American rights for Bepreve for the treatment of ocular allergy, various prostaglandin products and iganidipine to enhance ocular nerve blood flow.
In December 2009, we renegotiated our bromfenac license agreement with Senju to include, among other things, the expansion of our territory to include Canada and Mexico.
5
Table of Contents
Generally, under the terms of our agreements with Senju, we are responsible for all costs associated with developing the licensed products in ophthalmology for the United States and, with respect to Xibrom, Bepreve, prostaglandins and iganidipine, North America, including clinical trials, regulatory filings, manufacturing, and, if the product is approved, marketing and sales activities.
We have paid to Senju non-refundable milestone payments of $4 million relating to the development process and regulatory approval of both Istalol and Xibrom and are required to pay royalties on product sales.
We have paid to Senju non-refundable milestone payments of $4 million relating to the development process and regulatory approval of Bepreve and are required to pay royalties and milestones on product sales and product sales level achievements.
We will be required to pay to Senju non-refundable milestone payments of up to $3 million, some of which have been paid, if all such milestones relating to the development process and regulatory approval of ecabet sodium are accomplished, and royalties on future product sales.
We will be required to pay Senju non-refundable milestone payments of approximately $8 million, some of which have been paid, if all such milestones relating to the development process and regulatory approval of iganidipine are accomplished, and royalties on future product sales.
We will be required to pay Senju non-refundable milestone payments of approximately $8 million, some of which have been paid, if all such milestones relating to the development process and regulatory approval of a prostaglandin product are accomplished, and royalties on future product sales.
The license agreements with Senju for bromfenac, iganidipine and prostaglandins will terminate upon the later of (i) the last-to-expire licensed patent and (ii) ten years after the first commercial sale of the applicable licensed product. The license agreement with Senju for Istalol will terminate upon the last-to-expire licensed patent. The license agreements with Senju for ecabet sodium and Bepreve will terminate ten years after the later of (i) the last-to-expire licensed patent and (ii) ten years after the first commercial sale of the applicable licensed product.
Bepotastine Nasal Agreement With Mitsubishi Tanabe
In September 2007, we entered into a license agreement with Mitsubishi Tanabe Pharma Corporation under which we were granted exclusive North American rights to nasal dosage forms of bepotastine, an investigational product for the treatment of allergic rhinitis. We also obtained the right to develop other nasal bepotastine products, including a fixed combination with a steroid and a future right to negotiate for a North American license to oral dosage forms of bepotastine for allergy treatment.
Generally, under the terms of our agreement with Mitsubishi Tanabe, we are responsible for all costs associated with developing the licensed products in ophthalmology for the United States and, with respect to bepotastine nasal, North America, including clinical trials, regulatory filings, manufacturing, and, if the product is approved, marketing and sales activities.
Under the terms of our bepotastine nasal agreement with Mitsubishi Tanabe, we are required to pay Mitsubishi Tanabe non-refundable milestone payments of approximately $9 million, some of which have been paid, if all such milestones relating to the development process and regulatory approval of bepotastine nasal are accomplished, and royalties on future product sales.
The license agreement with Mitsubishi Tanabe for bepotastine nasal will terminate upon the later of (i) the last-to-expire licensed patent and (ii) ten years after the first commercial sale of the applicable licensed product.
Japan- Otsuka
In December 2001, we entered into certain agreements with Otsuka Pharmaceutical Co., Ltd., or Otsuka, with respect to the commercialization of Vitrase in Japan for ophthalmic uses in the posterior region of the eye. Under the terms of our agreements with Otsuka, Otsuka is responsible for preclinical studies, clinical trials, applying for and obtaining regulatory approvals and other development activities for Vitrase for ophthalmic uses in the posterior region of the eye in Japan.
In September 2009, we modified our existing License and Supply Agreements with Otsuka. Among other changes, the Supply Agreement terminated, resulting in us having no future obligation to supply Otsuka with hyaluronidase for injection. As a result, we recognized $3.1 million of previously deferred income primarily related to the termination of that supply agreement.
6
Table of Contents
Marketing and Sales
We continue to expand our commercial infrastructure in connection with the marketing, sale and distribution of our approved products in the United States. In late 2009 and early 2010, we expanded our sales force to a total of approximately 165 sales representatives to support our growing commercial activities.
InVentiv Pharma Services LLC provides us with administrative and other services, including training, analytics, and operational support. We target our commercialization efforts towards ophthalmologists, optometrists and allergists, who are the most prolific prescribers of ophthalmic beta-blockers, anti-inflammatories and allergy medications.
Customers and Distribution
We sell our approved products primarily to drug wholesalers, retailers and distributors, including large chain drug stores, hospitals, clinics, government agencies and managed healthcare providers such as health maintenance organizations and other institutions. These customers comprise a significant part of the distribution network for pharmaceutical products in the United States. This distribution network is continuing to undergo significant consolidation marked by mergers and acquisitions among wholesale distributors and the growth of large retail drug store chains. As a result, a small number of large, wholesale distributors control a significant share of the market, and the number of independent drug stores and small drug store chains has decreased. We expect that consolidation of drug wholesalers and retailers will, on an increasing basis, impact the net sales and gross margins of drug manufacturers and will create other competitive pressures.
We have engaged Cardinal Health PTS, LLC, or Cardinal Health, through its Specialty Pharmaceutical Services group, to act as our exclusive distributor for commercial shipment and distribution of our products to our customers in the United States. In addition to distribution services, Cardinal Health provides us with other related services, including product storage, returns, customer support, and administrative support.
Sales to AmeriSource Bergen Corp., McKesson HBOC and Cardinal Health, Inc. accounted for 17%, 40% and 35%, respectively, of our annual net revenues during 2009. The loss of any of these customers could materially and adversely affect our business, results of operations, financial condition and cash flows. Due to the relatively short lead-time required to fill orders for our products, backlog of orders is not material to our business.
Seasonality
We expect to experience seasonality with respect to sales of our ocular allergy products, specifically Bepreve. We expect larger sales in the spring through late summer and fewer sales in the late fall and winter.
In addition, although our ophthalmic pharmaceutical business is not materially affected by seasonal factors, we have noticed a historical trend with respect to sales. Specifically, our sales have tended to be lower during the first calendar quarter than the preceding fourth quarter.
Competition
The markets for therapies that treat diseases and conditions of the eye are subject to intense competition and technological change. Many companies, including major pharmaceutical companies, specialty pharmaceutical companies and specialized biotechnology companies, are engaged in activities similar to ours. Such companies include Allergan, Inc., Alcon Laboratories, Inc., Amphastar Pharmaceuticals, Inc., Bausch & Lomb, Incorporated, Johnson & Johnson, Novartis AG, Pfizer, Inc., and Inspire Pharmaceuticals, Inc. Many of these companies have substantially greater financial and other resources, larger research and development staffs and more extensive marketing and manufacturing organizations than ours.
Numerous companies are working on alternate therapies for ocular inflammation and pain, glaucoma, allergy, dry eye syndrome, ocular infection and other disease states of the eye.
In addition, competition from generic drug manufacturers is a major challenge in the United States to branded drug companies, like ISTA, and may have a material adverse effect on our product net revenues.
In January 2009, the patent on Xibrom expired, exposing us to potential future generic competition. Most companies that manufacture drug substances file a DMF with the FDA. Most companies that file ANDAs to gain approval to market a generic version of a previously patented pharmaceutical product will reference a DMF for the drug substance contained in the product. In July 2009, a new DMF was filed for bromfenac, the drug substance in Xibrom. Based upon publicly available information on the average FDA review times for ANDAs, we do not anticipate a generic form of bromfenac to gain approval by a competitor until 2011, or later.
7
Table of Contents
In October 2004, the FDA granted Istalol a “BT” rating, which means that prescriptions for Istalol cannot be substituted legally at pharmacies with generic timolol maleate products. Nonetheless, we believe that certain pharmacies may have substituted, and may be continuing to substitute, Istalol prescriptions with generic timolol maleate solutions. We have completed an educational campaign to make our customers and pharmacies aware of Istalol’s BT rating and that Istalol cannot be substituted legally at pharmacies with generic timolol maleate products.
Manufacturing
We have a supply agreement with Senju for bepotastine besilate, which is the active pharmaceutical ingredient in Bepreve. Currently, Senju is our sole source for bepotastine besilate. The active ingredient for Xibrom is also supplied to us under an exclusive agreement from a sole supplier. We have supply agreements with Bausch & Lomb to manufacture commercial quantities of Istalol, Xibrom and Bepreve. Currently, Bausch & Lomb is our sole source for all three products.
Ovine hyaluronidase, the active pharmaceutical ingredient used in Vitrase, is processed in several stages to produce a highly purified raw material for formulation. We had a supply agreement with Biozyme Laboratories, Ltd., or Biozyme, for ovine hyaluronidase. Our supply agreement with Biozyme ended in August 2007. We are developing and are awaiting qualification from the FDA for an onsite hyaluronidase manufacturing facility. We anticipate receiving qualification of the manufacturing facility by the FDA during the first half of 2010. Our success in validating the manufacturing site for production of ovine hyaluronidase cannot be assured. We have a supply agreement with Alliance Medical Products, Inc. to manufacture commercial quantities of Vitrase 200 USP units/mL in sterile solution. Currently, Alliance Medical Products, Inc. is our sole source for Vitrase.
Research and Development
Since our inception, we have made substantial investments in research and development. During the years ended December 31, 2009, 2008 and 2007, we spent $24.9 million, $32.4 million and $32.5 million, respectively, on research and development activities.
We plan to focus our near-term research and development efforts on the later-stage products in our product development pipeline. Building on these development efforts, our goal is to continue our growth as a commercial stage, multi-specialty ophthalmology and allergy pharmaceutical company by developing or acquiring complementary products, either already marketed or in late-stage development. Some licensed or acquired products may require additional research and development activities prior to regulatory approval and commercialization.
Patents and Proprietary Rights
Our success depends in part on our ability to obtain patent protection for our inventions, to preserve our trade secrets and to operate without infringing the proprietary rights of third parties. Our strategy is to actively pursue patent protection in the United States and foreign jurisdictions for technology that we believe to be proprietary and that offers a potential competitive advantage. As of December 31, 2009, we owned 14 issued U.S. patents, 6 pending U.S. patent applications, 37 issued foreign patents, and 9 pending foreign patent applications. In addition, as of December 31, 2009, we licensed 6 issued U.S. patents, 3 pending U.S. patent applications, 1 issued foreign patent, and 1 pending foreign patent application.
The table below sets forth, for each of our material products or product candidates covered by a patent, the technology or technologies dependent on each such patent, the jurisdiction where such patent protection has been obtained, the expiration date of such patent, and whether we own or license such patent.
| Product or Product Candidate Subject to Patent Protection |
Technology |
Jurisdiction | Expiration Date |
Owned or Licensed Patent | ||||||||||
| Bepreve |
Bepotastine active ingredient | U.S. | 12/26/2016 | Licensed | ||||||||||
| Bepreve |
Formulation | U.S. | patent application | Licensed | ||||||||||
| Istalol |
Method of use | U.S. | 11/2/2018 | Licensed | ||||||||||
| Bromfenac dry eye |
Formulation and method of use | U.S. | patent applications | Owned and licensed | ||||||||||
| T-Pred |
Formulation and method of use | U.S. and Canada | patent application | Owned | ||||||||||
| Ecabet sodium |
Method of use | U.S. | 11/13/2016 | Licensed |
8
Table of Contents
In addition to patents, we rely on trade secrets and proprietary know-how. We seek protection of these trade secrets and proprietary know-how, in part, through confidentiality and proprietary information agreements. We make efforts to require our employees, directors, consultants and advisors, outside scientific collaborators and sponsored researchers, other advisors and other individuals and entities to execute confidentiality agreements upon the start of employment, consulting or other contractual relationships with us. These agreements provide that all confidential information developed or made known to the individual or entity during the course of the relationship is to be kept confidential and not be disclosed to third parties, except in specific circumstances. In the case of employees and some other parties, the agreements provide that all inventions conceived by the individual will be our exclusive property. These agreements may not provide meaningful protection for, or adequate remedies to protect, our technology in the event of unauthorized use or disclosure of information. Furthermore, our trade secrets may otherwise become known to, or be independently developed by, our competitors.
We have not conducted an extensive search of patents issued to other parties and no assurance can be given that such patents do not exist, have not been filed, or could not be issued which contain claims relating to our technology and products. If such patents do exist, the owners may bring claims against us for infringement, which may have an adverse effect on our business.
We also file trademark applications to protect the names of our products. These applications may not mature to registration and may be challenged by third parties. In addition, some of our trademarks, including Xibrom, are owned by, or assignable to, our licensors, such as Senju, and upon expiration or termination of the license agreements, we may no longer be able to use these trademarks.
Government Regulation
Our pharmaceutical products are subject to extensive government regulation in the United States. If we ever decide to distribute our products abroad, our products would also be subject to extensive foreign government regulation. In the United States, the FDA regulates pharmaceutical products. FDA regulations govern the testing, manufacturing, advertising, promotion, labeling, sale and distribution of our products.
In general, the FDA approval process for drugs includes, without limitation:
| • | preclinical studies; |
| • | submission of an investigation new drug, or IND, application for clinical trials; |
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of the product; |
| • | submission of an NDA to obtain marketing approval; |
| • | review of the NDA; and |
| • | inspection of the facilities used in the manufacturing of the drug to assess compliance with the FDA’s current Good Manufacturing Practice, or cGMP, regulations. |
Preclinical studies include laboratory evaluation of the product, as well as animal studies to assess the potential safety and efficacy of the product. These studies must be performed according to good laboratory practices. The results of the preclinical studies, together with manufacturing information and analytical data, are submitted to the FDA as part of the IND application. Clinical trials may begin 30 days after the IND application is received, unless the FDA raises concerns or questions about the conduct of the clinical trials. If concerns or questions are raised, the IND application sponsor and the FDA must resolve any outstanding concerns before clinical trials can proceed.
We cannot assure you that submission of an IND application for any of our product candidates will result in authorization to commence clinical trials. Clinical trials involve the administration of the product that is the subject of the trial to volunteers or patients under the supervision of a qualified principal investigator. Each clinical trial must be reviewed and approved by an independent institutional review board at each institution at which the study will be conducted. The institutional review board will consider, among other things, ethical factors, safety of human subjects and the possible liability of the institution. Also, clinical trials must be performed according to good clinical practices. Good clinical practices are enumerated in FDA regulations and guidance documents.
Clinical trials typically are conducted in three sequential phases: Phases I, II and III, with Phase IV studies sometimes required to be conducted after approval. Drugs for which Phase IV studies are required include those approved under accelerated approval regulations. The four phases may overlap. In Phase I clinical trials, the drug is usually tested on a small number of healthy volunteers to determine:
| • | safety; |
9
Table of Contents
| • | any adverse effects; |
| • | proper dosage; |
| • | absorption; |
| • | metabolism; |
| • | distribution; |
| • | excretion; and |
| • | other drug effects. |
In Phase II clinical trials, the drug is usually tested on a limited number of subjects (generally up to several hundred subjects) to preliminarily evaluate the efficacy of the drug for specific, targeted indications, determine dosage tolerance and optimal dosage, and identify possible adverse effects and safety risks.
In Phase III clinical trials, the drug is usually tested on a larger number of subjects (up to several thousand), in an expanded patient population and at multiple clinical sites. The FDA may require that we suspend clinical trials at any time on various grounds, including if the FDA makes a finding that the subjects are being exposed to an unacceptable health risk.
Following successful conclusion of the Phase III clinical trial, an NDA is submitted to the FDA. The NDA must include comprehensive and complete descriptions of the preclinical testing, clinical trials, and the chemical manufacturing and control requirements of a drug that enable the FDA to determine the drug’s safety and efficacy. An NDA must be approved by the FDA before any of our drugs can be marketed commercially in the United States.
The FDA testing and approval process requires substantial time, effort and money. We cannot assure you that any NDA we submit for our product candidate will be timely approved, if ever.
In Phase IV clinical trials or other post-approval commitments, additional studies and patient follow-up are conducted to gain experience from the treatment of patients in the intended therapeutic indication. Additional studies and follow-up are also conducted to document a clinical benefit where drugs are approved under accelerated approval regulations and based on surrogate endpoints. In clinical trials, surrogate endpoints are alternative measurements of the symptoms of a disease or condition that are substituted for measurements of observable clinical symptoms. Failure to promptly conduct Phase IV clinical trials and follow-up could result in expedited withdrawal of products approved under accelerated approval regulations.
The facilities, procedures, and operations of our contract manufacturers must be determined to be adequate by the FDA before product approval. Manufacturing facilities are subject to inspections by the FDA for compliance with cGMP, licensing specifications, and other FDA regulations before and after an NDA has been approved. Foreign manufacturing facilities are also subject to periodic FDA inspections or inspections by foreign regulatory authorities. Among other things, the FDA may withhold approval of NDAs or other product applications of a facility if deficiencies are found at the facility. Vendors that supply us finished products or components used to manufacture, package and label products are subject to similar regulation and periodic inspections.
Following such inspections, the FDA may issue notices on Form 483 and Warning Letters that could cause us to modify certain activities identified during the inspection. A Form 483 notice is generally issued at the conclusion of an FDA inspection and lists conditions the FDA investigators believe may violate cGMP or other FDA regulations. FDA guidelines specify that a Warning Letter be issued only for violations of “regulatory significance” for which the failure to adequately and promptly achieve correction may be expected to result in an enforcement action.
In addition, the FDA imposes a number of complex regulatory requirements on entities that advertise and promote pharmaceuticals, including, but not limited to, standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities, and promotional activities involving the Internet.
Failure to comply with FDA and governmental regulations can result in fines, unanticipated compliance expenditures, recall or seizure of products, total or partial suspension of production and/or distribution, suspension of the FDA’s review of NDAs, injunctions, disqualification from participation in government reimbursement programs and criminal prosecution. Any of these actions could have a material adverse effect on us. For clinical trials conducted outside the United States, the clinical stages are generally comparable to the phases of clinical development established by the FDA.
10
Table of Contents
In the United States, physicians, hospitals and other healthcare providers that purchase pharmaceutical products generally rely on third-party payors, principally private health insurance plans, Medicare and, to a lesser extent, Medicaid, to reimburse all or part of the cost of the product and procedure for which the product is being used. Even if a product is approved for marketing by the FDA, there is no assurance that third-party payors will cover the cost of the product and related medical procedures. Although they are not required to do so, private health insurers often follow the Medicare program’s lead when determining whether or not to reimburse for a drug. To support our applications for reimbursement coverage with Medicare and other major third-party payors, we intend to use data from clinical trials. The lack of satisfactory reimbursement for our drug products would limit their widespread use and lower potential product net revenues.
Our interactions with physicians and other healthcare professional are subject to both federal and state law and regulation designed to prohibit companies from wrongfully inducing physicians and others from prescribing and using our products. We have adopted a comprehensive compliance program to regulate our personnel’s interactions with physicians and others, to attempt to comply with these regulations.
Federal, state and local laws of general applicability, such as laws regulating working conditions, also govern us. In addition, we are subject to various federal, state and local environmental protection laws and regulations, including those governing the discharge of material into the environment. We do not expect the costs of complying with such environmental provisions to have a material effect on our earnings, cash requirements or competitive position in the foreseeable future.
Human Resources
As of January 31, 2010, we had 315 full-time employees. Of our employees, 51 are engaged in research and development, 8 in manufacturing, 17 in quality assurance and quality control, 209 in sales and marketing, and 30 in administration and finance. Our employees do not have a collective bargaining agreement. We consider our relations with our employees to be good.
General Information
We incorporated in California in February 1992 as Advanced Corneal Systems, Inc. In March 2000, we changed our name to ISTA Pharmaceuticals, Inc., and we reincorporated in Delaware in August 2000. Our corporate headquarters and principal research laboratories are located at 15295 Alton Parkway, Irvine, CA 92618, and our telephone number is (949) 788-6000.
We make the following reports available on our website, at www.istavision.com, free of charge as soon as practicable after filing with the U.S. Securities and Exchange Commission, or SEC:
| • | our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to these reports; |
| • | our policies related to corporate governance, including our Code of Ethics and Conduct which apply to our directors, officers and employees (including our principal executive officer and principal financial and accounting officer) that we have adopted to meet the requirements set forth in the rules and regulations of the SEC and its corporate governance principles; and |
| • | the charters of the Audit, Compensation and Nominating & Corporate Governance Committees of our Board of Directors. |
All such reports are also available free of charge via EDGAR through the SEC website at www.sec.gov. In addition, the public may read and copy materials filed by ISTA with the SEC at the SEC’s public reference room located at 100 F St., NE, Washington, D.C., 20549. Information regarding operation of the SEC’s public reference room can be obtained by calling the SEC at 1-800-SEC-0330.
| Item 1A | Risk Factors |
In addition to other information included in this Annual Report on Form 10-K, the following factors, among others, could cause actual results to differ materially from those contained in forward-looking statements contained in this Annual Report on Form 10-K, and thus should be considered carefully in evaluating our business and future prospects. The following risk factors are not an exhaustive list of the risks associated with our business. New factors may emerge or changes to these risks could occur that could materially affect our business.
Risks Related to Our Business
If we do not timely receive and maintain regulatory approvals for our products or product candidates, we will not be able to commercialize our products, which would substantially impair our ability to generate revenues and materially harm our business and financial condition.
11
Table of Contents
Approval from the FDA is necessary to manufacture and market pharmaceutical products in the United States. Four of our products, Xibrom, Istalol, Bepreve and Vitrase, have received regulatory approval from the FDA. We currently have filed for approval for XiDay, our once-daily formulation of bromfenac, the active pharmaceutical ingredient in Xibrom.
The regulatory approval process is extensive, time-consuming and costly, and the FDA may not approve additional product candidates, or the timing of any such approval may not be appropriate for our product launch schedule and other business priorities, which are subject to change.
FDA approval of our products can be delayed, limited or not granted for many reasons, including, among others:
| • | FDA officials may not find a product candidate safe or effective to merit an approval; |
| • | FDA officials may not find that the data from preclinical testing and clinical trials justifies approval, or they may require additional studies that would make it commercially unattractive to continue pursuit of approval; |
| • | the FDA might not approve the processes or facilities of our contract manufacturers or raw material suppliers or our manufacturing processes or facilities; |
| • | the FDA may change its approval policies or adopt new regulations; and |
| • | the FDA may approve a product candidate for indications with labeling claims that are narrow or that place our product at a competitive disadvantage, which may limit our sales and marketing activities or otherwise adversely impact the commercial potential of a product. |
If the FDA does not approve our product candidates in a timely fashion with suitable labeling claims, or we terminate development of any of our product candidates due to difficulties or delays encountered in clinical testing and the regulatory approval process, it may have a material adverse impact on our business, results of operations and financial condition.
If clinical trials for our drug candidates are unsuccessful or delayed, we will be unable to meet our anticipated development and commercialization timelines, which could harm our business and financial condition.
Clinical testing of pharmaceutical products required to obtain approval is also a long, expensive and uncertain process. Even if initial results of preclinical studies or clinical trial results are positive, we may obtain different results in later stages of drug development, including failure to show desired safety and efficacy. The clinical trials of any of our product candidates could be unsuccessful, which would prevent us from obtaining regulatory approval and commercializing the product. If we terminate development of any of our product candidates due to difficulties or delays encountered in clinical testing, it may have a material adverse impact on our business, results of operations and financial condition.
The success of any of our product acquisition and licensing activities are subject to uncertainty and any completed acquisitions or licenses may reduce our earnings, be difficult to integrate, not perform as expected or require us to obtain additional financing.
We regularly evaluate selective acquisitions and look to continue to enhance our product line by acquiring rights to additional products and compounds. Such acquisitions may be carried out through the purchase of assets, joint ventures and licenses or by acquiring other companies. However, we cannot assure you that we will be able to complete acquisitions or in-licensing arrangements that meet our target criteria on satisfactory terms, if at all. Successfully integrating a product acquisition or in-licensing arrangement can be a lengthy and complex process. The diversion of our management's attention and any delays or difficulties encountered in connection with any of our acquisitions or arrangements could result in the disruption of our ongoing business or inconsistencies in standards, controls, procedures and policies that could negatively affect our ability to maintain relationships with customers, suppliers, employees and others with whom we have business dealings. In addition, other companies, including those with substantially greater resources than ours, may compete with us for the acquisition of product or in-licensing candidates and approved products, resulting in the possibility that we devote resources to potential acquisitions or arrangements that are never completed. If we do engage in any such acquisition or arrangement, we will incur a variety of costs, and we may never realize the anticipated benefits of the acquisition or arrangement in light of those costs. If we fail to realize the expected benefits from acquisitions or arrangements we may consummate in the future, whether as a result of unidentified risks, integration difficulties, regulatory setbacks or other events, our business, results of operations and financial condition could be adversely affected.
In addition, our product acquisition and licensing activities may require us to obtain additional debt or equity financing, resulting in increased debt obligations or dilution of ownership to our existing stockholders, as applicable. Therefore, we may not be able to finance acquisitions on terms satisfactory to us, if at all.
12
Table of Contents
If generic manufacturers obtain approval for generic versions of our products, our business, results of operations and financial condition may suffer.
Under the Federal Food, Drug and Cosmetics Act, or the FDCA, the FDA can approve an ANDA for a generic version of a branded drug, and under what is referred to as a Section 505(b)(2) NDA for a modified version of an existing branded drug, without the ANDA applicant undertaking the clinical testing necessary to obtain approval to market a new drug. In place of such clinical studies, an ANDA applicant usually needs only to submit data demonstrating that its product has the same active ingredient(s) and is bioequivalent to the branded product, in addition to any data necessary to establish that any difference in strength, dosage form, inactive ingredients, or delivery mechanism does not result in different safety and efficacy profiles, as compared to the branded drug.
The FDCA requires an applicant for a drug that relies, at least in part, on the existing approval of one of our branded drugs, to notify us of their application and potential infringement of our patent rights. Upon receipt of this notice, we have 45 days to bring a patent infringement suit in federal district court against the company seeking approval of a product covered by one of our patents. The discovery, trial and appeals process in such suits can take several years. If such a suit is commenced, the FDCA provides a 30-month stay on the FDA’s approval of the competitor’s application. Such litigation is often time-consuming and quite costly and may result in generic competition if such patent(s) are not upheld or if the generic competitor is found not to infringe such patent(s). If the litigation is resolved in favor of the applicant or the challenged patent expires during the 30-month stay period, the stay is lifted and the FDA may thereafter approve the application based on the standards for approval of ANDAs and Section 505(b)(2) NDAs.
In January 2009, the patent on Xibrom expired, and we lost regulatory exclusivity for Xibrom. Because of the patent expiration, competitors became eligible to receive approval for ANDAs for ophthalmic formulations of bromfenac with indications identical to Xibrom. During the second quarter of 2008, we submitted a Citizen Petition to the FDA requesting that the FDA refrain from approving any applications that contain bromfenac manufactured outside the United States until the FDA conducts full inspections and determines that manufacturers are in compliance with applicable regulations. The FDA denied such Citizen Petition in the fourth quarter of 2008. During the second quarter of 2008, we submitted a second Citizen Petition to the FDA requesting that the FDA refrain from approving any application for generic bromfenac ophthalmic formulations unless specific standards are satisfied, including but not limited to, corneal safety standards. On December 15, 2008, the FDA notified us that it had not yet reached a decision on the second Citizen Petition and that further review and analysis was required. The FDA published a guidance document in the Federal Register in January 2009 outlining its interpretation of responding to Citizens Petitions as required under the Food and Drug Administration Amendments Act that was passed in September 2007. In the guidance document, the FDA noted that it would not be required to respond to Citizens Petitions within the 180-day period from when the petition was filed in the event that an ANDA had not been filed as of the petition filing date. Because of this guideline, we have concluded that no ANDA was on file with the FDA for Xibrom as of June 23, 2008. During the fourth quarter of 2008, we submitted a third Citizen Petition to the FDA requesting that the FDA refrain from approving any applications for ophthalmic pharmaceutical products that do not contain standards for endotoxin levels and water for injection. On June 2, 2009, the FDA denied our petition. On June 30, 2009, we filed a Petition for Reconsideration of the denial of our Petition. On December 28, 2009, we received a letter from FDA informing us that the FDA has not reached a determination on our Petition for Reconsideration. We cannot predict whether the FDA will grant or deny our two pending Citizen Petitions or when it may take action with respect to such Citizen Petitions. In July 2009, a DMF was filed for bromfenac. Based upon publicly available information on the average FDA review times for ANDAs, we do not anticipate a generic form of bromfenac to gain approval by a competitor until 2011, or later.
The filing of any ANDA or Section 505(b)(2) NDAs with respect to Xibrom, or any of our other branded drugs, could have an adverse impact on our net revenues and results of operations. In 2009, Xibrom accounted for approximately 73% of our net revenues and 77% of our gross profit. Moreover, if the patents covering our branded drugs (other than Xibrom) were not upheld in litigation or if a competitor is found not to infringe these patents, the resulting competition would have a material adverse effect on our business, results of operations and financial condition.
If our products do not gain market acceptance, our business will suffer.
A number of factors may affect the market acceptance of our products or any other products we develop or acquire, including, among others:
| • | the price of our products relative to other therapies for the same or similar treatments; |
| • | the perception by patients, physicians and other members of the health care community of the safety and efficacy of our products for their prescribed treatments; |
| • | the availability of satisfactory levels, or at all, of third party reimbursement for our products and related treatments; |
| • | our ability to fund our sales and marketing efforts; and |
| • | the effectiveness of our sales and marketing efforts. |
13
Table of Contents
In addition, our ability to market and promote our products is restricted to the indications and labeling claims approved by the FDA. If the approved labeling is restrictive, our sales and marketing efforts and market acceptance and the commercial potential of our products may be negatively affected.
If our products do not gain market acceptance, we may not be able to fund future operations, including the development or acquisition of new product candidates and/or our sales and marketing efforts for our approved products, which would cause our business to suffer.
If we fail to properly manage our anticipated growth, our business could suffer.
Rapid growth of our business is likely to place a significant strain on our managerial, operational and financial resources and systems. To manage our anticipated growth successfully, we must attract and retain qualified personnel and manage and train them effectively. We are dependent on our personnel and third parties to effectively manufacture, market, sell and distribute our products. We will also continue to depend on our personnel and third parties to successfully develop and acquire new products. Further, our anticipated growth will place additional strain on our suppliers and manufacturers, resulting in an increased need for us to carefully manage these relationships and monitor for quality assurance. If we do not grow as we expect, if we fail to manage our growth effectively or if we do not develop and expand a successful commercial infrastructure, our business and financial results will be materially harmed.
We have a history of net losses and negative cash flow, and we may need to raise additional working capital.
We have never been profitable and we might never become profitable. As of December 31, 2009, our accumulated deficit was $397.3 million (including a non-cash valuation warrant adjustment of $52.1 million), including a net loss of approximately $57.8 million for the year ended December 31, 2009. As of December 31, 2009, we had approximately $53.7 million in cash and cash equivalents and working capital of $29.1 million. We believe our current cash and cash equivalents on hand, together with borrowings available under our revolving credit facility with Silicon Valley Bank, or Revolving Credit Facility, and other borrowing arrangements, will be sufficient to finance anticipated capital and operating requirements for at least the next twelve months.
If we are unable to generate sufficient product net revenues, we may be required to raise additional capital in the future through collaborative agreements or public or private equity or debt financings. If we are required to raise additional capital in the future such additional financing may not be available on favorable terms, or at all, or may be dilutive to our existing stockholders. In addition, our 2008 facility agreement with certain institutional investors, which we refer to as the Facility Agreement, and our Revolving Credit Facility contain restrictions on our ability to incur certain indebtedness without the prior consent of our lenders. If we fail to obtain additional capital as and when required such failure could have a material adverse impact on our business, results of operations and financial condition.
Adverse economic conditions may have material adverse consequences on our business, results of operations and financial condition.
Unpredictable and unstable changes in economic conditions, including recession, inflation, increased government intervention, or other changes, may adversely affect our general business strategy. If the current equity and credit markets further deteriorate, or do not continue to improve, it may make any necessary debt or equity financing more difficult, more costly, and more dilutive. While we believe we have adequate capital resources to meet current working capital and capital expenditure requirements, a radical economic downturn, a double-dip recession, or an increase in our expenses could require additional financing on less than attractive rates or on terms that are excessively dilutive to existing stockholders. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and stock price and could require us to delay or abandon clinical development plans or plans to acquire additional technology.
These economic conditions not only limit our access to capital, but also make it difficult for our customers and us to accurately forecast and plan future business activities, and they could cause businesses to slow spending on our products and services, which would delay and lengthen sales cycles. Furthermore, during challenging economic times, our customers may face issues gaining timely access to sufficient credit, which could result in an impairment of their ability to make timely payments to us. In addition, the recent economic crisis could also adversely impact our suppliers’ ability to provide us with materials and components, either of which may negatively impact our business, financial condition and results of operations. There is a risk that one or more of our current suppliers may encounter difficulties during challenging economic times, which would directly affect our ability to attain our operating goals.
14
Table of Contents
If we are required to immediately repay our outstanding borrowings, our financial position could be negatively impacted.
Outstanding amounts under our Revolving Credit Facility with Silicon Valley Bank bear interest at variable rates, which may expose us to interest rate risk. If interest rates increase, our debt service obligations on the variable rate indebtedness would increase and our income and cash flows would decrease. The loan and security agreement related to the Revolving Credit Facility also contains certain covenants based on our financial performance. If we violate any of these financial performance covenants, or are otherwise in default, our lender has the option to declare all outstanding borrowings immediately due and payable, which could also cause a default under our Facility Agreement, thereby allowing the lenders under our Facility Agreement to accelerate the payment of the amounts outstanding thereunder. In that event, we may not have sufficient resources to pay the outstanding amounts and would need to obtain additional financing, which may not be available on reasonable terms or at all.
If actual future payments or credits for allowances, discounts, product returns, rebates, chargebacks and other discounts, such as wholesaler fees, exceed the estimates we made at the time of the sale of our products, our financial position, results of operations and cash flows may be materially and negatively impacted.
We recognize revenues from product sales when there is persuasive evidence that an arrangement exists, when title has passed, the price is fixed or determinable, and we are reasonably assured of collecting the resulting receivable. We recognize product revenues net of estimated allowances for discounts, product returns, rebates, chargebacks and other discounts, such as wholesaler fees. If actual future payments for allowances for discounts, product returns, wholesaler fees, rebates and chargebacks exceed the estimates we made at the time of sale, our business, results of operations and financial condition would be negatively impacted.
In general, we are obligated to accept from our customers the return of pharmaceutical products that have reached their expiration date. We authorize returns for damaged products and, on a limited basis, exchanges for expiring and expired products in accordance with our return goods policy and procedures, and have established reserves for such amounts at the time of sale. We typically refund the agreed portion of the sales price by the issuance of a credit, rather than cash refund or exchanges for inventory, and the returned product is destroyed. With the launch of each of our products, we recorded a sales return allowance, which was larger for stocking orders than subsequent re-orders. To date, actual product returns have not exceeded our estimated allowances for returns. Although we believe that our estimates and assumptions are reasonable as of the date when made, actual results may differ significantly from these estimates. Our business, results of operations and financial condition may be materially and negatively impacted if actual returns exceed our estimated allowances for returns.
Customers typically process their claim for allowances such as early pay discounts promptly, usually within the established payment terms. We monitor actual credit memos issued to our customers and compare such actual amounts to the estimated provisions, in the aggregate, for each allowance category to assess the reasonableness of the various reserves at each balance sheet date. Differences between our estimated allowances and actual credits issued have not been significant, and are accounted for in the current period as a change in estimate in accordance with generally accepted accounting principles. Our business, results of operations and financial condition may be materially and negatively impacted if actual credits issued exceed our estimated allowances for such credits.
Our quarterly results may fluctuate significantly and could fall below the expectations of securities analysts and investors, resulting in a decline in our stock price.
Our quarterly operating results may fluctuate significantly because of several factors, including:
| • | the level of our net revenues, expenses and gross margins; |
| • | the volatility of our stock price and its impact on the valuation of our warrants and other financial instruments; |
| • | the timing of our regulatory submissions or approvals, or the failure to receive regulatory approvals; |
| • | the initiation and progress of our clinical trials and other product development activities; |
| • | the introduction of competitive products, including potential generic products, and announcements from competitors regarding actual or potential products under development or new commercial products, and the impact of competitive products and pricing; |
| • | the level of orders within a given quarter and preceding quarters; |
| • | the service fees charged and the levels of inventory for our products maintained by our customers, including wholesalers; |
| • | the timing of our product shipments and our customer’s receipt of such shipments within a given quarter; |
15
Table of Contents
| • | the timing of introducing new products; |
| • | the changes in our pricing policies or in the pricing policies of our competitors or suppliers; and |
| • | our product mix and dependence on a small number of products for most of our net revenues. |
We expect to experience seasonality with respect to sales of our ocular allergy products, specifically Bepreve. We expect larger sales in the spring through late summer and fewer sales in the late fall and winter. In addition, although our ophthalmic pharmaceutical business is not materially affected by seasonal factors, we have noticed a historical trend with respect to sales. Specifically, our sales have tended to be lower during the first calendar quarter than the preceding fourth quarter. Due to these and other factors, we believe that quarter-to-quarter comparisons of results from operations, or any other similar period-to-period comparisons, should not be construed as reliable indicators of our future performance. In any quarterly period, our results may be below the expectations of market analysts and investors, which would likely cause the trading price of our common stock to decrease.
Our partners may terminate, or fail to perform their duties under our agreements, in which case our ability to commercialize our products may be significantly impaired.
We have entered into licensing agreements with Senju relating to Istalol, Xibrom, Bepreve, ecabet sodium, iganidipine, and certain prostaglandin compounds, including latanoprost. With respect to Xibrom, Bepreve, ecabet sodium and iganidipine, certain patent and other intellectual property rights we have received from Senju have been licensed to Senju from third parties. As a result, Senju’s license of such rights to us is subject to Senju maintaining and performing its obligations under these third party license agreements. We have also entered into an exclusive licensing agreement with Mitsubishi Tanabe Pharma Corporation, from whom we obtained the North American rights to nasal (including intranasal) dosage forms of bepotastine. Certain intellectual property rights we received from Mitsubishi Tanabe have been licensed to Mitsubishi Tanabe from a third party, and thus Mitsubishi Tanabe’s license of such rights to us is subject to Mitsubishi Tanabe maintaining and performing its obligations under such third party license agreement.
Any termination of our agreements by our partners could delay or stop the development and/or commercialization of our product candidates. In addition, any failure by Senju or Mitsubishi Tanabe to perform their respective obligations under their license agreements with third parties, or any adverse modification or termination of these third party license agreements, could significantly impair our ability to continue or stop our development and/or commercialization of any product candidates or products for which Senju or Mitsubishi Tanabe has licensed us rights subject to these third party agreements. Our agreements with Senju and Mitsubishi Tanabe generally contain reciprocal terms providing that neither we nor they may develop products that directly compete in the same form with the products involved in the agreement. Nonetheless, our partners may develop competing products in different forms or products that compete indirectly with our products.
If we are unable to obtain materials from our sole source suppliers in a timely manner or our sole source suppliers do not meet their commitments, our product development and commercialization efforts for our product candidates could be delayed or stopped.
We utilize third-party manufacturers and suppliers for the manufacture and supply of our products and for their active and other ingredients. These third-party manufacturers and suppliers are subject to extensive regulation by the FDA and other agencies, and we do not have control over their compliance with these regulations. The disqualification of these manufacturers and suppliers through their failure to comply with regulatory requirements could negatively impact our business because the delays and costs in obtaining and qualifying alternate suppliers (if such alternative suppliers are available, which they may not be) could delay clinical trials or otherwise inhibit our ability to bring approved products to market, which could have a material adverse effect on our business, results of operations and financial condition.
Some materials used in our products are currently obtained from a single source. We have a supply agreement with Senju for bepotastine besilate, which is the active pharmaceutical ingredient in Bepreve. Currently, Senju is our sole source for bepotastine besilate. The active ingredient for Xibrom is also supplied to us under an exclusive agreement from a sole supplier. We also have supply agreements with Bausch & Lomb to manufacture commercial quantities of Istalol, Xibrom and Bepreve, and, currently, Bausch & Lomb is our sole source supplier for such products. Hyaluronidase, our active ingredient in Vitrase, was manufactured by Biozyme. Our supply agreement with Biozyme ended in August 2007. We are developing and are awaiting qualification from the FDA for an onsite hyaluronidase manufacturing facility. We anticipate receiving qualification of the manufacturing facility by the FDA during the first half of 2010. Our success in validating the manufacturing site for production of ovine hyaluronidase cannot be assured. We have a supply agreement with Alliance Medical Products, Inc. to manufacture commercial quantities of Vitrase 200 USP units/mL in sterile solution. Currently, Alliance Medical Products is our sole source supplier for Vitrase.
16
Table of Contents
We have not established and may not be able to establish arrangements with additional suppliers for certain of these ingredients or products. Difficulties in our relationships with our suppliers or delays or interruptions in such suppliers’ supply of our requirements could limit or stop our ability to provide sufficient quantities of our product candidates on a timely basis for clinical trials and, for our approved products, could limit or stop commercial sales, which would have a material adverse effect on our business, results of operations and financial condition.
Our dependence upon key personnel to operate our business puts us at risk of a loss of expertise if key personnel were to leave us.
We depend upon the experience and expertise of our executive management team. The competition for executives, as well as for skilled product development, marketing and sales, and technical personnel, in the pharmaceutical industry is intense and we may not be able to retain or recruit the personnel we need. If we are not able to attract and retain existing and additional highly qualified management, sales, clinical and technical personnel, we may not be able to successfully execute our business strategy.
Risks Related to Our Industry
Compliance with extensive government regulations or other third parties to which we are subject is expensive and time consuming, and may result in the delay, cessation or cancellation of product sales, introductions or modifications.
Extensive industry regulation has had, and will continue to have, a significant impact on our business. All pharmaceutical companies, including us, are subject to extensive, complex, costly and evolving regulation by the federal government, principally the FDA, and foreign and state government agencies. The FDCA, the Controlled Substances Act and other domestic and foreign statutes and regulations govern or influence the testing, manufacturing, packing, labeling, storing, record keeping, safety, approval, advertising, promotion, sale and distribution of our products. Under certain of these regulations, we and our contract suppliers and manufacturers are subject to periodic inspection of our or their respective facilities, procedures and operations and/or the testing of our products by the FDA and other authorities, which conduct periodic inspections to confirm that we and our contract suppliers and manufacturers are in compliance with all applicable regulations. The FDA also conducts pre-approval and post-approval reviews and plant inspections to determine whether our systems, or our contract suppliers’ and manufacturers’ processes, are in compliance with cGMP regulations and other FDA regulations.
We are dependent on maintaining FDA and other governmental approvals in order to manufacture, market, sell and ship our products. Consequently, there is always a risk that the FDA or other applicable governmental authorities will take post-approval action limiting, modifying or revoking our ability to manufacture or sell our products, or that the cost of maintaining such approvals will adversely affect our results of operations. Certain of the FDA’s policies and procedures are under review by new leadership and it is uncertain whether any changes arising from such review could adversely affect our products and business.
We currently have certain raw materials manufactured in foreign countries and the manufacturers of those materials are subject to regulation and inspection by both the FDA and local governmental authorities. We may also elect in the future to market certain of our products manufactured, in foreign countries which would require further approvals by local governmental authorities.
To the extent that our products are reimbursed by Medicare, Medicaid or other federal programs, our marketing and sales activities will be subject to federal regulation. Federal agencies in recent years have initiated investigations against, and entered into multi-million dollar settlements with, a number of pharmaceutical companies alleging violations of fraud and abuse provisions. We will need to ensure that our sales force is properly trained to comply with these laws. Even with such training, there is a risk that some of our marketing practices could come under scrutiny, or that we will not be able to institute or continue certain marketing practices. The majority of states also have statutes or regulations similar to these federal laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Sanctions under these federal and state laws may include civil monetary penalties, exclusion of a manufacturer’s products from reimbursement under government programs, criminal fines and imprisonment. Because of the breadth of these laws and the narrowness of the safe harbors, it is possible that some of our business activities could be subject to challenge under one or more of such laws.
In addition, the FDA imposes a number of complex regulatory requirements on entities that advertise and promote pharmaceutical products, including, but not limited to, standards and regulations for direct-to-consumer advertising, off-label promotion, industry sponsored scientific and educational activities, and promotional activities involving the Internet.
17
Table of Contents
In April 2008, we received subpoenas from the office of the U.S. Attorney for the Western District of New York requesting the production of documents regarding promotional, educational and other activities relating to Xibrom. We are cooperating with the government to provide the requested documents. At this time, we are unable to predict the outcome of this matter or reasonably estimate the amount or range of amounts of fines or penalties that might result from an adverse outcome. From April 2008 through December 31, 2009, we incurred approximately $2.8 million in legal fees responding to the document requests and expect to incur significant expenses in the future. If, as a result of its review of the requested documents and other evidence, the government chooses to engage in civil litigation or initiate a criminal prosecution against us, we could have to expend significant resources to defend such action and, if not successful, incur or pay substantial fines or penalties, including but not limited to monetary settlements, disqualification from government reimbursement programs, or entering into a corporate integrity agreement with the government, any of which could have a material adverse effect on our business, results of operations and financial condition.
Our interactions with physicians and other healthcare professionals are subject to both federal and state law and regulation designed to prohibit companies from wrongfully inducing physicians and others from prescribing and using our products. We have adopted a comprehensive compliance program to regulate our personnel’s interactions with physicians and others, to attempt to comply with these regulations. However, because of the breadth of these laws and regulations and subjective nature of their fundamental bases, it is possible that some of our business activities could be subject to challenge under one or more of such laws.
If our past or present operations are found to be in violation of any of the laws described above or other similar governmental regulations to which we are subject, we may be subject to the applicable penalty associated with the violation which could adversely affect our ability to operate our business, results of operations and financial condition.
We may be required to initiate or defend against legal proceedings related to intellectual property rights, which may result in substantial expense, delay and/or cessation of our development and commercialization of our products.
We rely on patents to protect our intellectual property rights. The strength of this protection, however, is uncertain. For example:
| • | others may claim that our patents and pending patent applications cover products and/or technology that we did not invent first; |
| • | others may claim that we were not the first to file patent applications for these inventions; |
| • | others may independently develop similar or alternative technologies or duplicate our technologies; |
| • | our pending patent applications may not result in issued patents; and |
| • | any of our issued patents, or pending patent applications that result in issued patents, will be held invalid or not infringed in the event the patents are asserted against others. |
As of December 31, 2009, we owned 14 issued U.S. patents, 6 pending U.S. patent applications, 37 issued foreign patents, and 9 pending foreign patent applications. In addition, as of December 31, 2009, we licensed 6 issued U.S. patents, 3 pending U.S. patent applications, 1 issued foreign patent, and 1 pending foreign patent application. Our existing patents, or any patents issued to us as a result of such applications, may not provide us a basis for commercially viable products, may not provide us with any competitive advantages, or may face third-party challenges or be the subject of further proceedings limiting their scope or enforceability. We may become involved in interference proceedings in the U.S. Patent and Trademark Office to determine the priority of our inventions. In addition, costly litigation could be necessary to protect our patent position. We license patent rights from Senju related to Istalol, Xibrom, Bepreve, ecabet sodium, iganidipine and certain prostaglandin compounds, including latanoprost. We also license patent rights from Mitsubishi Tanabe for bepotastine in nasal dosage form. Some of these license agreements do not permit us to control the prosecution, maintenance, protection and/or defense of such patents. If the licensor chooses not to protect and enforce its own patent rights, we may not be able to take actions to secure our related product marketing rights. In addition, if such patent licenses are terminated before the expiration of the licensed patents, we may no longer be able to continue to manufacture and sell these products covered by the patents. In this regard, certain patent rights licensed from Senju and Mitsubishi Tanabe were licensed by them from third parties. As a result, any failure by Senju or Mitsubishi Tanabe to perform their respective obligations under their license agreements with third parties, or any adverse modification or termination of these third party license agreements, could significantly impair our ability to continue or stop our development and/or commercialization of any product candidates or products for which Senju and Mitsubishi Tanabe have licensed us rights subject to these third party license agreements.
We also rely on trade secrets, unpatented proprietary know-how and continuing technological innovation that we seek to protect with confidentiality agreements with employees, consultants and others with whom we discuss our business. Disputes may arise concerning the ownership of intellectual property or the applicability or enforceability of these agreements, and we might not be able to resolve these disputes in our favor.
18
Table of Contents
We also rely on trademarks to protect the names of our products. These trademarks may be challenged by others. If we enforce our trademarks against third parties, such enforcement proceedings may be expensive. Some of our trademarks, including Xibrom, are owned by, or assignable to, our licensors, and upon expiration or termination of the applicable license agreements, we may no longer be able to use these trademarks.
In addition to protecting our own intellectual property rights, we may be required to defend against third parties who assert patent, trademark or copyright infringement or other intellectual property claims against us based on what they believe are their own intellectual property rights. We may be required to pay substantial damages, including but not limited to treble damages, for past infringement if it is ultimately determined that our products infringe a third party’s intellectual property rights. Even if infringement claims against us are without merit, defending a lawsuit takes significant time, may be expensive and may divert management’s attention from other business concerns. Further, we may be stopped from developing, manufacturing or selling our products until we obtain a license from the owner of the relevant technology or other intellectual property rights. If such a license is available at all, it may require us to pay substantial royalties or other fees.
We have not conducted an extensive search of patents issued to other parties and such patents which contain claims relating to our technology and products may exist, may have been filed, or could be issued. If such patents do exist, the owners may bring claims against us for infringement, which might have an adverse effect on our business, results of operations and financial condition.
If third-party reimbursement is not available at satisfactory levels or at all, our products may not be accepted in the market.
Market acceptance of our products depends in part on the extent to which reimbursement for our products, and for our competitors’ products, and related treatments will be available from government health administration authorities, private health insurers, managed care organizations and other healthcare providers. Both governmental and private third-party payors are increasingly attempting to limit both the coverage and the level of reimbursement of new drug products to contain costs.
Any of our products that have been, or in the future are, approved by the FDA may be purchased or reimbursed by state and federal government authorities, private health insurers and other organizations, such as health maintenance organizations and managed care organizations. Such third party payors increasingly challenge pharmaceutical product pricing. The trend toward managed healthcare in the United States, the growth of such organizations, and various legislative proposals and enactments to reform healthcare and government insurance programs, including the Medicare Prescription Drug Modernization Act of 2003, could significantly influence the manner in which pharmaceutical products are prescribed and purchased, resulting in lower prices and/or a reduction in demand. Such cost-containment measures and healthcare reforms could adversely affect our ability to sell our products. Furthermore, individual states have become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access, importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third party payors or other restrictions could negatively and materially impact our net revenues and financial condition. Similar regulatory and legislative issues are present in most other countries outside of the United States.
The U.S. Department of Defense’s, or DoD’s, TRICARE Retail Pharmacy program pursuant to section 703 of the National Defense Authorization Act of 2008, became effective on May 26, 2009. This regulation would require manufacturers to pay rebates to DoD on products distributed to TRICARE beneficiaries through retail pharmacies retroactive to January 28, 2008. The regulation requires that pharmaceuticals paid for by the DoD through the TRICARE Retail Pharmacy program be subject to the Federal Ceiling Price program, which would require manufacturers to provide DoD with a refund on pharmaceuticals utilized through the TRICARE Retail Pharmacy program. We have requested a waiver of the retroactive rebate for TRICARE Retail Pharmacy utilization for the period from January 28, 2008 to May 26, 2009. In addition, the regulation is currently the subject of litigation, and it is our belief that the retroactive application of the regulation is contrary to established case law. We have determined that payment of the retroactive rebate created by the regulation is neither reasonably estimable nor probable as of December 31, 2009. However, our financial position, results of operations and cash flows may be materially and negatively impacted if we are required to pay the retroactive rebate created by this regulation.
It is uncertain how any other policies and new healthcare legislation supported by the current presidential administration may impact the government and other third party payors’ reimbursement policies. Consequently, significant uncertainty exists as to the reimbursement status of healthcare products. Third-party payors may not establish adequate levels of reimbursement for any of our approved products or products we develop or acquire in the future, which could limit their market acceptance and result in a material adverse effect on our business, results of operations and financial condition.
19
Table of Contents
Continuing consolidation of our distribution network and the concentration of our customer base could adversely affect our results of operations.
Our principal customers are wholesale drug distributors and major retail drug store chains. These customers comprise a significant part of the distribution network for pharmaceutical products in the United States. This distribution network is continuing to undergo significant consolidation marked by mergers and acquisitions among wholesale distributors and the growth of large retail drug store chains. As a result, a small number of large wholesale distributors control a significant share of the market, and the number of independent drug stores and small drug store chains has decreased. We expect that consolidation of drug wholesalers and retailers could continue, likely resulting in increased service fees charged to drug companies and increase other competitive pressures on drug manufacturers. For the year ended December 31, 2009, our three largest customers, AmeriSource Bergen Corp., McKesson HBOC and Cardinal Health, Inc., accounted for 17%, 40% and 35%, respectively, of our net revenues. In addition, none of our customers are party to any long-term supply agreements with us which would enable them to change suppliers freely should they wish to do so. The loss of any of our customers could materially adversely affect our business, results of operations and financial condition.
We face intense competition and rapid technological change that could result in the development of products by others that are superior to the products we are developing.
We have numerous competitors in the United States and abroad, including major pharmaceutical and specialized biotechnology firms, universities and other research institutions that may be developing competing products. Our competitors include, among others, Allergan, Inc., Alcon Laboratories, Inc., Amphastar Pharmaceuticals, Inc., Bausch & Lomb, Incorporated, Johnson & Johnson, Novartis AG, Pfizer, Inc., and Inspire Pharmaceuticals, Inc. These competitors may develop technologies and products that are more effective or less costly than our current or future products or product candidates or that could render our technologies, products and product candidates obsolete or noncompetitive. Many of these competitors have substantially more resources and product development, manufacturing and marketing experience and capabilities than we do. Many of our competitors also have more resources committed to, and expertise in, effectively commercializing, marketing, and promoting products approved by the FDA, including communicating the efficacy, safety and value of the products to actual and prospective customers and medical professionals. In addition, many of our competitors have significantly greater experience than we do in undertaking preclinical testing and clinical trials of pharmaceutical product candidates and obtaining FDA and other regulatory approvals of products and therapies for use in healthcare.
We are exposed to product liability claims, and insurance against these claims may not be available to us on reasonable terms, or at all.
The design, development, manufacture and sale of our products involve an inherent risk of product liability claims by consumers and other third parties. As a commercial company, we may be subject to various product liability claims. In addition, we may in the future recall or issue field corrections related to our products due to manufacturing deficiencies, labeling errors or other safety or regulatory reasons. We may experience material losses due to product liability claims, product recalls or corrections. These events, among others, could result in additional regulatory controls, such as the performance of costly post-approval clinical studies or revisions to our approved labeling that could limit the indications or patient population for our products or could even lead to the withdrawal of a product from the market. Furthermore, any adverse publicity associated with such an event could cause consumers to seek alternatives to our products, which may cause our sales to decline, even if our products are ultimately determined not to have been the primary cause of the event.
We currently maintain sold products and clinical trial liability insurance with per occurrence and aggregate coverage limits of $10 million. The coverage limits of our insurance policies may be inadequate to protect us from any liabilities we might incur in connection with clinical trials or the sale of our products. Product liability insurance is expensive and in the future may not be available on commercially acceptable terms, or at all. A successful claim or claims brought against us in excess of our insurance coverage could materially harm our business, results of operations and financial condition.
Risks Related to Our Stock
Our stock price is subject to significant volatility.
Since 2004, the daily closing price per share of our common stock has ranged from a high of $15.05 per share to a low of $0.36 per share. Our stock price has been and may continue to be subject to significant volatility. Among others, the following factors may cause the market price of our common stock to fall:
| • | the scope, outcome and timeliness of any governmental, court or other regulatory action that may involve us, including, without limitation, the scope, outcome or timeliness of any product approval, inspection or other action of the FDA; |
20
Table of Contents
| • | market acceptance and demand for our approved products; |
| • | the availability to us, on commercially reasonable terms or at all, of third-party sourced products and materials; |
| • | timely and successful implementation of our strategic initiatives, including the expansion of our commercial infrastructure to support the marketing, sale, and distribution of our approved products; |
| • | developments concerning proprietary rights, including the ability of third parties to assert patents or other intellectual property rights against us which, among other things, could cause a delay or disruption in the development, manufacture, marketing or sale of our products; |
| • | competitors’ publicity regarding actual or potential products under development or new commercial products, and the impact of competitive products, including potential generic products, and pricing; |
| • | period-to-period fluctuations in our financial results; |
| • | future sales of debt or equity securities by us; |
| • | sales of our securities by our directors, officers or significant stockholders; |
| • | availability of capital from hedge funds, mutual funds and others; |
| • | comments made by securities analysts; and |
| • | economic and other external factors, including disasters and other crises. |
We participate in a highly dynamic industry, which often results in significant volatility in the market price of our common stock irrespective of company performance. Fluctuations in the price of our common stock may be exacerbated by conditions in the healthcare and technology industry segments or conditions in the financial markets generally.
If our stock does not continue to be traded on an established exchange, an active trading market may not develop and the trading price of our stock may decline.
Our common stock is listed on The NASDAQ Global Market. In order to maintain that listing, we are required to satisfy minimum financial and continued listing requirements, including, without limitation, maintaining a $1.00 per share minimum closing bid price for our common stock. During the last twelve months, our closing stock price has ranged from $0.73 to $6.00 per share.
If the closing bid price of our common stock is below $1.00 for 30 consecutive business days, we could receive notice from NASDAQ stating that the minimum bid price for our common stock is below continuing listing standards. If our common stock were threatened with delisting from The NASDAQ Global Market, we may, depending on the circumstances, seek to extend the period for regaining compliance with NASDAQ listing requirements by moving our common stock to The NASDAQ Capital Market, or we may pursue other strategic alternatives to meet the continuing listing standards. For example, if appropriate, we may request approval by our stockholders to implement a reverse stock split in order to regain compliance with NASDAQ’s minimum bid price requirement.
If our common stock is delisted by NASDAQ, our common stock may be eligible to trade on the NYSE Alternext U.S., the OTC Bulletin Board or the Pink OTC Markets. In such an event, it could become more difficult to dispose of, or obtain accurate quotations for the price of, our common stock, and there would likely also be a reduction in our coverage by security analysts and the news media, which could cause the price of our common stock to decline further. In addition, if we are not listed on a national securities exchange, it would constitute an event of default under our Facility Agreement, which would also trigger a default under our Revolving Credit Facility, unless we renegotiate our Facility Agreement.
Trading in our stock over the last twelve months has been limited, so investors may not be able to sell as much stock as they want at prevailing prices.
The average daily trading volume in our common stock for the year ended December 31, 2009 was approximately 185,000 shares and the average daily number of transactions was approximately 663 for the same period. If limited trading in our stock continues, it may be difficult for investors to sell their shares in the public market at any given time at prevailing prices. Moreover, the market price for shares of our common stock may be made more volatile because of the relatively low volume of trading in our common stock. When trading volume is low, significant price movement can be caused by the trading in a relatively small number of shares. Volatility in our common stock could cause stockholders to incur substantial losses.
21
Table of Contents
Substantial future sales of our common stock in the public market may depress our stock price and make it difficult for investors to recover the full value of their investment in our shares.
We have approximately 33.3 million shares of common stock outstanding, most of which are freely tradable. In addition, as of December 31, 2009, an aggregate of 7.1 million shares of common stock were issuable upon exercise of outstanding options, 15.0 million shares of common stock are issuable upon the exercise of certain warrants issued under the Facility Agreement and 6.9 million shares remain available for issuance under our equity incentive plans. The market price of our common stock could decrease due to sales of a large number of shares or the perception that such sales could occur. These factors also could make it more difficult to raise funds through future offerings of common stock.
Our directors, officers and principal stockholders have significant voting power and may take actions that may not be in the best interests of our other stockholders.
As of December 31, 2009, our officers, directors and principal stockholders, each holding more than 5% of our common stock, beneficially own approximately 90% of our common stock in the aggregate. As a result, these stockholders, if they act together, will be able to control the management and affairs of our company and most matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. This concentration of ownership may have the effect of delaying or preventing a change in control and might adversely affect the market price of our common stock.
Our stockholder rights plan, provisions in our charter documents, and Delaware law may inhibit a takeover of us, which could limit the price investors might be willing to pay in the future for our common stock, and could entrench management.
We have a stockholder rights plan that may have the effect of discouraging unsolicited takeover proposals, thereby entrenching current management and possibly depressing the market price of our common stock. The rights issued under the stockholder rights plan would cause substantial dilution to a person or group that attempts to acquire us on terms not approved in advance by our board of directors. In addition, our charter and bylaws contain provisions that may discourage unsolicited takeover proposals that stockholders may consider to be in their best interests. These provisions include:
| • | a classified board of directors; |
| • | the ability of the board of directors to designate the terms of and issue new series of preferred stock; |
| • | advance notice requirements for nominations for election to the board of directors; and |
| • | special voting requirements for the amendment of our charter and bylaws. |
We are also subject to anti-takeover provisions under Delaware law, each of which could delay or prevent a change of control. Together these provisions and the stockholder rights plan may make more difficult the removal of management and may discourage transactions that otherwise could involve payment of a premium over prevailing market prices for our common stock.
22
Table of Contents
We do not anticipate declaring any cash dividends on our common stock.
We have never declared or paid cash dividends on our common stock and do not plan to pay any cash dividends in the near future. Our current policy is to retain all funds and any earnings for use in the operation and expansion of our business. The payment of cash dividends by us is restricted by our Facility Agreement, which contains restrictions prohibiting us from paying any cash dividends without the lender’s prior approval. If we do not pay dividends, our stock may be less valuable to investors because a return on their investment will only occur if our stock price appreciates.
| Item 1B: | Unresolved Staff Comments. |
On December 23, 2009, we received a comment letter from the staff of the SEC’s Department of Corporate Finance related to a review of our Annual Report on Form 10-K for the year ended December 31, 2008, Form 10-K/A for the year ended December 31, 2008, and Definitive Proxy Statement filed October 30, 2009. The SEC requested additional information and provided comments related to (i) certain of our disclosures pertaining to our patents and proprietary rights, (ii) our revenue recognition policies, (iii) our accounting treatment of warrants and derivatives, and (iv) certain of our disclosures pertaining to executive compensation. We responded to the SEC on January 29, 2010 and believe we have addressed the SEC’s comments relating to our patents and proprietary rights, our revenue recognition policies, and our accounting treatment of warrants and derivatives, in this filing. In addition, we will address the SEC’s comments relating to our executive compensation disclosures in an amendment to this Form 10-K to be filed not later than 120 days after the end of the fiscal year covered by this Form 10-K. However, as of the filing date of this Form 10-K, the SEC’s review of our filings is not complete, and they may have further comments.
| Item 2: | Properties. |
We do not own real property. We currently lease five facilities, which approximate 52,920 square feet of laboratory and office space, located at 15295, 15279, 15285 and two different suites at 15273 Alton Parkway in Irvine, California. The term of the 15295, 15279 and 15285 leases expire on December 31, 2010, and the term of the 15273 leases expire on March 31, 2016, but may be renewed by us for an additional five year term. We believe that these five facilities are adequate, suitable and of sufficient capacity to support our immediate needs. Additional space may be required, however, as we expand our research and clinical development, manufacturing and selling and marketing activities. We are currently exploring facility options, including leasing an alternate site or renewing our existing lease agreements.
| Item 3: | Legal Proceedings. |
In April 2008, we received subpoenas from the office of the U.S. Attorney for the Western District of New York requesting the production of documents regarding promotional, educational and other activities relating to Xibrom. We are cooperating with the government to provide the requested documents. At this time, we are unable to predict the outcome of this matter or reasonably estimate the amount or range of amounts of fines or penalties that might result from an adverse outcome. From April 2008 through December 31, 2009, we incurred approximately $2.8 million in legal fees responding to the document requests and expect to incur significant expenses in the future. If, as a result of its review of the requested documents and other evidence, the government chooses to engage in civil litigation or initiate a criminal prosecution against us, we could have to expend significant resources to defend such action and, if not successful, incur or pay substantial fines or penalties, including but not limited to monetary settlements, disqualification from government reimbursement programs, or entering into a corporate integrity agreement with the government, any of which could have a material adverse effect on our business, results of operations and financial condition.
We are involved in other legal proceedings incidental to our business from time to time. Except as described immediately above, we do not believe that pending actions or proceedings, either individually or in the aggregate, will have a material adverse effect on our financial condition, results of operations or cash flows, and adequate provision has been made for the resolution of such actions and proceedings.
| Item 4: | Submission of Matters to a Vote of Security Holders. |
Set forth below is information concerning each matter submitted to a vote at the Annual Meeting of Stockholders held on December 7, 2009.
Proposal No. 1: The stockholders elected two Class III directors to serve for a term of three years expiring upon the 2012 Annual Meeting of Stockholders or until his or her successor has been duly elected and qualified.
| Nominee |
Votes For | Votes Withheld | ||
| Dean J. Mitchell |
27,544,075 | 1,442,191 | ||
| Wayne I. Roe |
28,435,498 | 550,768 |
23
Table of Contents
Proposal No. 2: The stockholders ratified the appointment of BDO Seidman, LLP as our independent auditors for the fiscal year ended December 31, 2009 (with 28,821,768 votes for, 82,190 votes against, and 82,308 votes abstaining).
Proposal No. 3: The stockholders approved the amendments to the 2004 Performance Incentive Plan (with 17,190,236 votes for, 5,181,248 votes against, and 34,439 votes abstaining).
Proposal No. 4: The stockholders approved the 2009 Employee Stock Purchase Plan (with 18,893,367 votes for, 3,470,964 votes against, and 41,592 votes abstaining).
24
Table of Contents
PART II
| Item 5: | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Price Range of Common Stock
Our common stock is listed on The NASDAQ Global Market under the symbol “ISTA.” The following table shows the high and low sale prices for our common stock as reported by The NASDAQ Global Market during the calendar quarters indicated:
| High | Low | |||||
| Year Ended December 31, 2008 |
||||||
| First Quarter |
$ | 5.71 | $ | 1.16 | ||
| Second Quarter |
2.39 | 1.27 | ||||
| Third Quarter |
2.53 | 1.30 | ||||
| Fourth Quarter |
1.60 | 0.29 | ||||
| Year Ended December 31, 2009 |
||||||
| First Quarter |
$ | 2.24 | $ | 0.72 | ||
| Second Quarter |
5.53 | 1.70 | ||||
| Third Quarter |
6.83 | 3.85 | ||||
| Fourth Quarter |
4.88 | 3.31 | ||||
| Year Ending December 31, 2010 |
||||||
| First Quarter (through February 12, 2010) |
$ | 4.83 | $ | 3.41 | ||
Holders of Common Stock
As of January 31, 2010, there were approximately 131 stockholders of record of our common stock based upon the records of our transfer agent which do not include beneficial owners of common stock whose shares are held in the names of various securities brokers, dealers and registered clearing agencies.
Dividend Policy
We have never declared or paid any cash dividends on our common stock and do not intend to pay any cash dividends on our common stock in the foreseeable future. The payment of cash dividends by us is restricted by our Facility Agreement and our Revolving Credit Facility which contain restrictions prohibiting us from paying any cash dividends without the lenders’ prior consent.
Equity Compensation Plans
| Plan category |
(a) Number of securities to be issued upon exercise of outstanding options, warrants and rights |
(b) Weighted- average exercise price of outstanding options, warrants and rights |
(c) Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a) | ||||
| Equity compensation plans approved by security holders (1)(2) |
6,906,002 | $ | 5.57 | 6,917,871 | |||
| Equity compensation plans not approved by security holders (3)(4) |
15,145,461 | $ | 1.55 | — | |||
| Total |
22,051,463 | $ | 2.81 | 6,917,871 | |||
| (1) | Our 2000 Employee Stock Purchase Plan, or 2000 ESPP, previously provided for annual increases in the number of shares available for issuance under the plan on the first day of each year equal to the lesser of 20,000 shares, 1.5% of the outstanding shares of common stock on the first day of the year, or a lesser amount as the Board of Directors may determine. On December 7, 2009, the stockholders approved the 2009 Employee Stock Purchase Plan, or 2009 ESPP, with 3,000,000 shares initially reserved and an increase each January 1, beginning January 1, 2011, in the number of shares reserved by the lesser of (i) of 1% of our outstanding common stock or (ii) an amount determined by the Compensation Committee; however, in no event will the number of shares reserved exceed the lesser of 10% of our outstanding common stock or 5,000,000 shares. The initial offering period will commence on January 1, 2010 and end on June 30, 2010, with subsequent offering periods commencing on six-month intervals thereafter beginning on July 1, 2010. Following approval of the 2009 ESPP, we terminated the 2000 ESPP. |
25
Table of Contents
| (2) | On December 7, 2009, the stockholders approved the Fourth Amendment and Restatement of the 2004 Stock Plan, which increased the number of shares available by 6,000,000 shares to an aggregate of 12,153,107 shares, of which up to 1,450,000 shares may be issued in connection with restricted stock awards or performance share awards. |
| (3) | In December 2001, the Board of Directors granted our new Chief Executive Officer and President, as an inducement to his employment, a stand-alone option agreement to purchase 100,461 shares of our common stock for a purchase price of $20.00 per share. In June 2002, the Board of Directors granted our new Vice President, Sales & Marketing, as an inducement to his employment, a stand-alone option agreement to purchase 30,000 shares of our common stock of for a purchase price of $8.50. In August 2002, the Board of Directors granted our new Vice President, Operations, as an inducement to his employment, a stand-alone option agreement to purchase 15,000 shares of our common stock for a purchase price of $6.90. Each option holder may purchase up to 25% of the shares under each option on the first anniversary of the option grant date and the right to purchase the remaining shares vests in equal monthly installments so that each option is fully vested four years after the date of grant, except that the option granted to our Chief Executive Officer was amended to provide that 25,113 options would not be exercisable until December 22, 2010. |
| (4) | In 2008, in conjunction with our $65 million Facility Agreement, we issued warrants to purchase an aggregate of 15 million shares of our common stock at an exercise price of $1.41 per share. The warrants expire on September 26, 2014. |
Stock Price Performance Graph
The following graph compares our total cumulative stockholder return as compared to The NASDAQ Global Market and U.S. index, or NASDAQ U.S. Index, and the NASDAQ Pharmaceutical Index for the period beginning on December 31, 2004 and ending on December 31, 2009. Total stockholder return assumes $100.00 invested at the beginning of the period in our common stock, the stocks represented by the NASDAQ U.S. Index and the NASDAQ Pharmaceutical Index, respectively. Total return assumes reinvestment of dividends; we have paid no dividends on our common stock.
26
Table of Contents
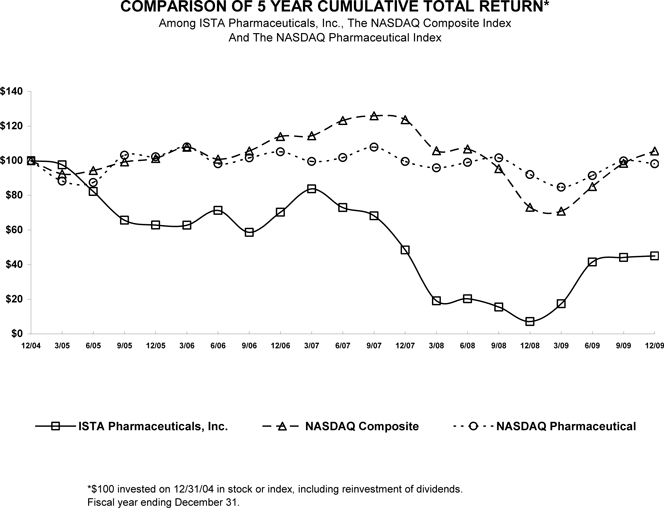
The material in the above performance graph does not constitute soliciting material and should not be deemed filed or incorporated by reference into any other Company filing, whether under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made on, before or after the date of this report and irrespective of any general incorporation language in such filing, except to the extent we specifically incorporate this performance graph by reference therein.
27
Table of Contents
| Item 6: | Selected Financial Data. |
The table below presents our selected consolidated financial data as of and for the years ended December 31, 2009, 2008, 2007, 2006 and 2005. The following selected consolidated financial data has been derived from our audited consolidated financial statements and should be read in conjunction with our consolidated financial statements contained herein, and related notes thereto, as well as our “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this Annual Report on Form 10-K.
| Years Ended December 31, (in thousands, except per share data) |
||||||||||||||||||||
| 2009 | 2008 (as adjusted) |
2007 (as adjusted) |
2006 (as adjusted) |
2005 | ||||||||||||||||
| Consolidated Statement of Operations Data: |
||||||||||||||||||||
| Revenues: |
||||||||||||||||||||
| Product sales, net |
$ | 107,593 | $ | 82,798 | $ | 58,589 | $ | 32,729 | $ | 10,382 | ||||||||||
| License revenue |
3,055 | 278 | 278 | 278 | 278 | |||||||||||||||
| Total revenues |
110,648 | 83,076 | 58,867 | 33,007 | 10,660 | |||||||||||||||
| Cost of products sold |
27,278 | 21,947 | 15,864 | 9,943 | 3,542 | |||||||||||||||
| Gross profit margin |
83,370 | 61,129 | 43,003 | 23,064 | 7,118 | |||||||||||||||
| Costs and expenses: |
||||||||||||||||||||
| Research and development |
24,904 | 32,400 | 32,492 | 23,826 | 16,611 | |||||||||||||||
| Selling, general and administrative |
56,377 | 53,539 | 46,603 | 37,357 | 30,599 | |||||||||||||||
| Total costs and expenses |
81,281 | 85,939 | 79,095 | 61,183 | 47,210 | |||||||||||||||
| Income (loss) from operations |
2,089 | (24,810 | ) | (36,092 | ) | (38,119 | ) | (40,092 | ) | |||||||||||
| Other (expense) income: |
||||||||||||||||||||
| Interest income |
— | 714 | 2,141 | 1,879 | 1,642 | |||||||||||||||
| Interest expense |
(8,591 | ) | (8,100 | ) | (7,669 | ) | (3,976 | ) | (30 | ) | ||||||||||
| Loss on extinguishment of debt |
— | (2,497 | ) | — | — | — | ||||||||||||||
| Gain (loss) on derivative valuation |
1,177 | 26 | (197 | ) | — | — | ||||||||||||||
| Loss on warrant valuation |
(52,066 | ) | — | — | — | — | ||||||||||||||
| Other, net |
(363 | ) | — | — | — | — | ||||||||||||||
| Net loss |
$ | (57,754 | ) | $ | (34,667 | ) | $ | (41,817 | ) | $ | (40,216 | ) | $ | (38,480 | ) | |||||
| Net loss per common share, basic and diluted |
$ | (1.74 | ) | $ | (1.05 | ) | $ | (1.41 | ) | $ | (1.55 | ) | $ | (1.51 | ) | |||||
| Shares used in computing net loss per common share, |
33,228 | 33,028 | 29,621 | 26,011 | 25,490 | |||||||||||||||
| As of December 31, (in thousands) |
||||||||||||||||||||
| 2009 | 2008 (as adjusted) |
2007 (as adjusted) |
2006 (as adjusted) |
2005 | ||||||||||||||||
| Consolidated Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 53,702 | $ | 53,016 | $ | 46,140 | $ | 38,934 | $ | 38,626 | ||||||||||
| Working capital |
29,113 | 31,500 | 32,686 | 27,998 | 32,990 | |||||||||||||||
| Total assets |
89,144 | 82,660 | 71,716 | 59,743 | 45,339 | |||||||||||||||
| Deferred income |
— | 3,055 | 3,333 | 3,611 | 3,994 | |||||||||||||||
| Convertible notes |
— | — | 40,253 | 40,000 | — | |||||||||||||||
| Facility agreement |
57,438 | 55,157 | — | — | — | |||||||||||||||
| Warrant liability |
58,663 | — | — | — | — | |||||||||||||||
| Other long-term obligations |
325 | 450 | 407 | 270 | 255 | |||||||||||||||
| Accumulated deficit |
(397,272 | ) | (343,243 | ) | (308,576 | ) | (266,759 | ) | (226,543 | ) | ||||||||||
| Total stockholders’ equity (deficit) |
(78,028 | ) | (17,199 | ) | 3,881 | 4,226 | 30,335 | |||||||||||||
28
Table of Contents
| Item 7: | Management’s Discussion and Analysis of Financial Condition and Results of Operations. |
This Annual Report on Form 10-K contains forward-looking statements that have been made pursuant to the provisions of the Private Securities Litigation Reform Act of 1995 and concern matters that involve risks and uncertainties that could cause actual results to differ materially from those projected in the forward-looking statements. Discussions containing forward-looking statements may be found in the material set forth under “Business,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and in other sections of this Form 10-K. Words such as “may,” “will,” “should,” “could,” “expect,” “plan,” “anticipate,” “believe,” “estimate,” “predict,” “potential,” “continue” or similar words are intended to identify forward-looking statements, although not all forward-looking statements contain these words. Although we believe that our opinions and expectations reflected in the forward-looking statements are reasonable as of the date of this Annual Report on Form 10-K, we cannot guarantee future results, levels of activity, performance or achievements, and our actual results may differ substantially from the views and expectations set forth in this Annual Report on Form 10-K. We expressly disclaim any intent or obligation to update any forward-looking statements after the date hereof to conform such statements to actual results or to changes in our opinions or expectations. Readers are urged to carefully review and consider the various disclosures made by us, which attempt to advise interested parties of the risks, uncertainties, and other factors that affect our business, set forth in detail in Item 1A of Part I, under the heading “Risk Factors.”
The following discussion and analysis should be read in conjunction with our consolidated financial statements and the related notes to those statements contained elsewhere in this Annual Report on Form 10-K.
Overview
We are a commercial stage, multi-specialty pharmaceutical company developing, marketing and selling our own products in the United States. We manufacture our products through third party contracts and we in-license or acquire promising new technology to add to our internal development efforts from time to time.
Our products and product candidates seek to treat serious diseases of the eye and allergies and include therapies for ocular inflammation and pain, glaucoma, dry eye and ocular and nasal allergies. The U.S. prescription markets our therapies seek to address include key segments of the $5.5 billion ophthalmic pharmaceutical market and the $2.2 billion nasal allergy market.
We currently have four products available for sale in the United States and Puerto Rico: Xibrom (bromfenac sodium ophthalmic solution) for the treatment of inflammation and pain following cataract surgery, Bepreve (bepotastine besilate ophthalmic solution) for the treatment of ocular itching associated with allergic conjunctivitis, Istalol (timolol maleate ophthalmic solution) for the treatment of glaucoma, and Vitrase (hyaluronidase for injection) for use as a spreading agent.
We have incurred losses since inception and had an accumulated deficit of $397.3 million (including a non-cash valuation warrant adjustment of $52.1 million) through December 31, 2009.
Results of Operations
Years Ended December 31, 2009, 2008 and 2007
Revenues. Net revenues were approximately $110.6 million in 2009, as compared to $83.1 million in 2008 and $58.9 million in 2007. The increase in revenues is the result of the following:
| • | Increased growth in prescription levels and market share for our core products, particularly for Xibrom; |
| • | Launch of our newest product, Bepreve, in September 2009, resulting in $1.7 million in net revenues; and |
| • | Recognition of $3.1 million in 2009 of previously deferred income primarily related to the termination of a supply agreement. |
29
Table of Contents
The following table sets forth our net revenues for each of our products for the years ended December 31, 2009, 2008 and 2007, respectively (dollars in millions):
| Years Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Xibrom |
$ | 81.1 | $ | 63.0 | $ | 42.1 | |||
| Istalol |
18.8 | 14.6 | 11.3 | ||||||
| Vitrase |
5.9 | 5.2 | 5.2 | ||||||
| Bepreve |
1.7 | — | — | ||||||
| Product sales, net |
107.5 | 82.8 | 58.6 | ||||||
| License revenue |
3.1 | 0.3 | 0.3 | ||||||
| Total revenues |
$ | 110.6 | $ | 83.1 | $ | 58.9 | |||
30
Table of Contents
Gross margin and cost of products sold. Gross margin for 2009 was 75%, or $83.4 million, as compared to 74%, or $61.1 million, for 2008 and 73%, or $43.0 million, for 2007. The increase in gross margin in 2009 as compared to 2008 and 2007 is primarily the result of continued increased growth in prescription levels and market share, particularly for Xibrom. Cost of products sold was $27.3 million in 2009, as compared to $21.9 million in 2008 and $15.9 million in 2007. Cost of products sold for the three years consisted primarily of standard costs for each of our commercial products, distribution costs, royalties, inventory reserves and other costs of products sold. The increase in costs of products sold during each of the three years is primarily the result of increased net product sales.
Research and development expenses. Research and development expenses were $24.9 million in 2009, $32.4 million in 2008 and $32.5 million in 2007. The decrease is research and development expenses in 2009, as compared to 2008, was primarily the result of a decrease in both clinical development costs, which include clinical investigator fees, study monitoring costs, data management costs, and manufacturing costs, offset by $3.0 million in milestone payments. The decrease in both clinical development costs and manufacturing development costs is due to the timing of clinical trials, both initiation and completion. During 2009, because the clinical trials had been completed previously and regulatory filings had been submitted to the FDA, we experienced a reduction in Bepreve expenses.
The 2007 research and development expenses included a one-time up-front payment to in-license Bepreve, which did not occur in 2008. Without giving effect to this one-time payment, the increase in research and development expenses in 2008 resulted from an increase in clinical development costs, which include FDA filing fees, clinical investigator fees, study monitoring costs and data management costs due the completion of the Bepreve Phase III clinical trial and ocular safety studies, additional clinical costs for XiDay and additional clinical costs for the ecabet sodium product.
Our research and development expenses to date have consisted primarily of costs associated with the clinical trials of our product candidates, compensation and other expenses for research and development personnel, costs for consultants and contract research organizations and costs related to the development of commercial scale manufacturing capabilities for Xibrom, Bepreve, Istalol and Vitrase.
Generally, our research and development resources are not dedicated to a single project but are applied to multiple product candidates in our portfolio. As a result, we manage and evaluate our research and development expenditures generally by the type of costs incurred. We generally classify and separate research and development expenditures into amounts related to clinical development costs, regulatory costs, pharmaceutical development costs, manufacturing development costs and medical affairs costs. In addition, we also record as research and development expenses any up-front and milestone payments that have been accrued to third parties prior to regulatory approval of a product candidate under our licensing agreements unless there is an alternative future use. In 2009, approximately 32% of our research and development expenditures were for clinical development costs, 20% were for regulatory costs, 6% were for pharmaceutical development costs, 12% were for manufacturing development costs, 13% were for medical affairs costs, 12% was for both a one-time milestone payment ($2.0 million) upon the FDA approval of our Bepreve NDA and a one-time milestone payment ($1.0 million) upon the FDA acceptance of our Bepreve NDA and approximately 5% for stock-based compensation costs ($1.2 million).
Changes in our research and development expenses in 2009 as compared to 2008 were primarily due to the following:
| • | Clinical Development Costs — Overall clinical costs, which include clinical investigator fees, study monitoring costs and data management, were $8.0 million for 2009 as compared to $17.0 million for 2008, or a decrease of $9.0 million. The decrease is due in part to the variation in the initiation, timing and completion of clinical studies year over year. In 2008, costs include those associated with our XiDay and our T-Pred studies and costs incurred in support of our Bepreve NDA. There was a significant reduction in 2009 of our clinical costs as the work on Bepreve was substantially completed. Additionally, we filed our Bepreve NDA in 2008 and received approval from the FDA to market Bepreve during the quarter ended September 30, 2009. |
| • | Regulatory Costs — Regulatory costs, which include compliance expenses for existing products and other activity for pipeline projects, were $5.0 million for 2009 as compared to $4.8 million for 2008. The increase of $0.2 million was due primarily to costs associated with the preparation of our sNDA filing for XiDay in December 2009 and our participation in an FDA advisory panel for Bepreve in June 2009. |
| • | Pharmaceutical Development Costs — Pharmaceutical development costs, which include costs related to the testing and development of our pipeline products, were $1.3 million in both 2009 and 2008. |
| • | Manufacturing Development Costs — Manufacturing development costs, which include costs related to production scale-up and validation, raw material qualification, and stability studies, were $3.1 million for 2009 as compared to $5.5 million for 2008, or a decrease of $2.4 million. The decrease is due primarily to a reduction in outside consulting costs and research related costs such as clinical supplies and stability studies. |
31
Table of Contents
| • | Medical Affairs Costs — Medical affairs costs, which include activities that relate to medical information in support of our products, were $3.3 million for 2009 as compared to $2.8 million for 2008. The increase of $0.5 million is primarily due to post-marketing studies related to our existing commercial products. |
In 2008, approximately 53% of our research and development expenditures were for clinical development costs, 15% were for regulatory costs, 4% were for pharmaceutical development costs, 17% were for manufacturing development costs, 8% were for medical affairs costs, and approximately 3% for stock-based compensation costs.
Changes in our research and development expenses in 2008 as compared to 2007 were primarily due to the following:
| • | Clinical Development Costs — Clinical development costs were $17.0 million for 2008 as compared to $13.6 million for 2007, or an increase of $3.4 million. The increase was primarily due to the continued efforts on the XiDay once-daily product, as well as the completion of the second Phase III clinical trial and ocular safety studies for Bepreve. |
| • | Regulatory Costs — Regulatory costs were $4.8 million for 2008 as compared to $5.0 million for 2007, or a decrease of $0.2 million. The decrease was primarily due to a reduction in regulatory activities. |
| • | Pharmaceutical Development Costs — Pharmaceutical development costs were $1.3 million for both 2008 and 2007. |
| • | Manufacturing Development Costs — Manufacturing development costs were $5.5 million for 2008 as compared to $5.9 million for 2007, or a decrease of $0.4 million. The decrease was primarily due to a reduction in research activities, specifically stability costs. |
| • | Medical Affairs Costs — Medical affairs costs were $2.8 million for 2008 as compared to $2.7 million for 2007, or an increase of $0.1 million. The increase was primarily due to post-marketing studies related to our existing commercial products. |
Our research and development activities reflect our efforts to advance our product candidates through the various stages of product development. The expenditures that will be necessary to execute our development plans are subject to numerous uncertainties, which may affect our research and development expenditures and capital resources. For instance, the duration and the cost of clinical trials may vary significantly depending on a variety of factors including a trial’s protocol, the number of patients in the trial, the duration of patient follow-up, the number of clinical sites in the trial, and the length of time required to enroll suitable patient subjects. Even if earlier results are positive, we may obtain different results in later stages of development, including failure to show the desired safety or efficacy, which could impact our development expenditures for a particular product candidate. Although we spend a considerable amount of time planning our development activities, we may be required to deviate from our plan based on new circumstances or events or our assessment from time to time of a product candidate’s market potential, other product opportunities and our corporate priorities. Any deviation from our plan may require us to incur additional expenditures or accelerate or delay the timing of our development spending. Furthermore, as we obtain results from trials and review the path toward regulatory approval, we may elect to discontinue development of certain product candidates in certain indications, in order to focus our resources on more promising candidates or indications. As a result, the amount or ranges of estimable cost and timing to complete our product development programs and each future product development program is not estimable.
Selling, general and administrative expenses. Selling, general and administrative expenses were $56.4 million in 2009, $53.5 million in 2008 and $46.6 million in 2007. The $2.9 million increase in 2009 as compared to 2008 was primarily attributable to higher sales and marketing expenses associated with launching Bepreve and expanding our sales force ($2.2 million), an overall increase in administrative costs, primarily legal expenses and personnel costs ($1.1 million), offset by a decrease in stock-based compensation costs ($0.4 million). Selling, general and administrative expenses are expected to increase in 2010 due to our sales force expansion.
The $6.9 million increase in 2008 as compared to 2007 primarily results from higher sales and marketing expenses ($3.3 million), an overall increase in administrative costs relating primarily to legal expenses ($3.1 million), and an increase in stock-based compensation costs ($0.5 million).
Stock-based compensation costs. Total stock-based compensation costs for the years ended December 31, 2009, 2008 and 2007 were $3.8 million, $4.0 million and $3.5 million, respectively. For the year ended December 31, 2009, we granted options to employees to purchase 1.3 million shares of common stock at a weighted average exercise price of $2.68 per share, equal to the fair market value of our common stock at the time of grant. In addition to stock options, we also issued restricted stock awards. Total stock-based compensation costs for the years ended December 31, 2009, 2008 and 2007 included $0.6 million, $0.6 million and $0.3 million, respectively, related to these restricted stock awards. The following table sets forth our stock-based compensation costs for the years ended December 31, 2009, 2008 and 2007, respectively (dollars in millions):
| Years Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Selling, general and administrative |
$ | 2.6 | $ | 3.0 | $ | 2.5 | |||
| Research and development |
1.2 | 1.0 | 1.0 | ||||||
| Share-based compensation costs |
$ | 3.8 | $ | 4.0 | $ | 3.5 | |||
32
Table of Contents
Interest income. Interest income was zero in 2009, $0.7 million in 2008 and $2.1 million in 2007. The decrease in interest income in 2009 was because our cash was not invested in interest bearing accounts as we sought to protect our principal. The decrease in interest income in 2008 was primarily attributable to the overall reduction in interest rates during 2008, lower cash on hand balances and other factors.
Interest expense. Interest expense was $8.6 million in 2009, $8.1 million in 2008 (as adjusted) and $7.7 million in 2007 (as adjusted). Interest expense in 2009 included interest payments on our Facility Agreement ($4.2 million), interest on our borrowings under our Revolving Credit Facility ($0.2 million), amortization of the deferred financing costs ($1.1 million), amortization of the derivative on the Facility Agreement ($0.4 million) and amortization of the discount on the Facility Agreement ($2.7 million). Included in 2008 and 2007 is the impact of the change in accounting principle as required by the Debt with Conversion and Other Options Topic of the FASB Accounting Standards Codification, which required retrospective application. This retrospective application required us to record additional non-cash interest expense of $2.7 million, or $0.08 per share, for the year ended December 31, 2008 and $3.6 million, or $0.12 per share for the year ended December 31, 2007. This additional non-cash interest expense represents the amortization of a debt discount recorded against our convertible notes on our balance sheet. Because our convertible notes were repaid in September 2008, there was no impact to 2009.
Loss on extinguishment of debt. The loss on extinguishment of debt of $2.5 million is due to the write-off of both the embedded derivative and the deferred financing costs related to the $40.0 million convertible notes in September 2008.
Gain (loss) on derivative valuation. Derivative valuation was a gain of $1.2 million in 2009, a gain of $26,000 in 2008 and a loss of $0.2 million in 2007. In 2009, the gain was the result of a decrease in the value of the derivative. During 2008, the derivative associated with the convertible notes issued in 2006 was written off, in connection with the repayment of the convertible notes, and the value of the derivative associated with the Facility Agreement increased, resulting in a net gain of $26,000. During 2007, the loss on the derivative valuation of $0.2 million was the result of an increase in the derivative value.
Loss on warrant valuation. Included in 2009 is the impact of the change in accounting principle as required by the Derivatives and Hedging Topic of the FASB Accounting Standards Codification, which required us to analyze the accounting for warrants under our Facility Agreement, issued in 2008. During 2008, the warrants were classified as equity. As a result of this change in accounting principle, we were required, due to certain provisions in the Facility Agreement, to reclassify the warrants as a liability. Specifically, because of the anti-dilutive provisions in the Warrant Agreement, where additional warrants might be issued should we issue additional equity, with such additional warrants being issued at a price equal to the fair value of the common stock being issued, but not less than $1.41, we were required to reclassify the warrants to a liability.
Additionally, the warrants will be marked to market and adjusted quarterly. We recorded a non-cash valuation adjustment for an additional $52.1 million, or $1.57 per share, in our financial results in 2009. The change in the valuation of the warrants was primarily driven by an increase in our stock price plus an increase in related volatility.
Income taxes. We incurred net operating losses for the years ended December 31, 2009, 2008 and 2007 and consequently did not pay any federal, state or foreign income taxes. At December 31, 2009, we had federal and state net operating loss carryforwards of approximately $178.9 million and $106.3 million, respectively, which we have fully reserved due to the uncertainty of realization. Our federal tax loss carryforwards will continue to expire in 2009, unless utilized. Our California tax loss carryforwards will begin to expire in 2012, unless utilized. We also have federal and California research tax credit carryforwards of approximately $9.7 million and $6.2 million, respectively. The federal research tax credits will begin to expire in 2010, unless utilized. Our California research tax credit carryforwards do not expire and will carryforward indefinitely until utilized.
2010 Financial Outlook
| • | We expect our net revenues for 2010 will be approximately $147 to $165 million. We continue to assume there will not be a generic to Xibrom in 2010. We expect net revenues from our Xibrom (including XiDay, assuming approval) franchise to be in the range of $95 to $105 million and anticipate net revenues from Bepreve will be at least $20 million. As in previous years, our net revenues are seasonal, with first quarter net sales typically being the lowest of the year and less than the prior quarter. |
33
Table of Contents
| • | We expect our gross margin in 2010 will be in the range of 74% to 76%. |
| • | We expect research and development expenses to be approximately 18 to 22% of net revenues, depending upon the progress of our clinical programs. In 2010, we will focus our development activities on our bromfenac as a treatment for dry eye disease and our nasal formulation of bepotastine for nasal allergies. We will initiate our full-scale Phase III clinical program in dry eye disease with bromfenac, as well as the long-term safety study required by the FDA in advance of an NDA filing. To maximize our bepotastine franchise, we will complete our formulation work for bepotastine nasal and initiate the clinical program with this product as a treatment for nasal allergies. |
| • | We expect selling, general and administrative expenses for 2010 to be approximately 48 to 52% of net revenues. |
| • | We expect our operating income for 2010 will be $8 to $10 million. |
| • | We expect our net income for 2010 will be at least $1 million, or earnings per share of at $0.02, excluding any mark-to-market adjustments relating to warrants. Once we are profitable, we expect our fully diluted common shares, including our outstanding shares of common stock, warrants and stock options on a treasury basis, will be approximately 49 million shares. |
| • | We expect our business to generate $6 to $10 million in cash in 2010. |
Liquidity and Capital Resources
As of December 31, 2009, we had approximately $53.7 million in cash and cash equivalents and working capital of $29.1 million. For the year ended December 31, 2009 we generated $2.7 million of positive cash flow. Historically, we have financed our operations primarily through sales of our debt and equity securities. Since March 2000, we have received gross proceeds of approximately $347.2 million from sales of our common stock and the issuance of promissory notes, warrants and debt.
Under our Revolving Credit Facility with Silicon Valley Bank, we may borrow up to the lesser of $25.0 million or 80% of eligible accounts receivable, plus the lesser of 25% of net cash or $10.0 million. As of December 31, 2009, we had $19 million available under the Revolving Credit Facility, of which we borrowed $13 million. All amounts borrowed under the Revolving Credit Facility were repaid in January 2010. All outstanding amounts under the Revolving Credit Facility bear interest at a variable rate equal to the lender’s prime rate plus a margin of 0.25%. In no event shall the interest rate on outstanding borrowings be less than 4.25%, which is payable on a monthly basis. The Revolving Credit Facility also contains customary covenants regarding operations of our business and financial covenants relating to ratios of current assets to current liabilities and is collateralized by all of our assets. An event of default under the Revolving Credit Facility will occur if, among other things, (i) we are delinquent in making payments of principal or interest on the Revolving Credit Facility; (ii) we fail to cure a breach of a covenant or term of the Revolving Credit Facility; (iii) we make a representation or warranty under the Revolving Credit Facility that is materially inaccurate; (iv) we are unable to pay our debts as they become due, certain bankruptcy proceedings are commenced or certain orders are granted against us, or we otherwise become insolvent; (v) an acceleration event occurs under certain types of other indebtedness outstanding from time to time. If an event of default occurs, the indebtedness to Silicon Valley Bank could be accelerated, such that it becomes immediately due and payable. As of December 31, 2009, we were in compliance with all of the covenants under the Revolving Credit Facility. Unless repaid earlier, all amounts owing under the Revolving Credit Facility will become due and payable on December 30, 2010. While we believe we will be able extend our agreement with Silicon Valley Bank upon its maturity or refinance outstanding amounts with another lender, we may not be able to do so due to general economic conditions surrounding the current crisis. If we are unable to renew our Revolving Credit Facility or obtain suitable alternative debt financing, it may adversely affect our ability to execute on our business plan.
In September 2008, we entered into a Facility Agreement with certain institutional accredited investors, which we refer to as the Lenders, to loan us up to $65.0 million. We borrowed the entire $65 million available under the Facility Agreement and issued warrants to purchase 15 million shares of common stock at an exercise price of $1.41 per share. Outstanding amounts drawn under the Facility Agreement accrue interest at a fixed rate of 6.5% per annum, payable quarterly in cash in arrears. We are required to repay the Lenders 33% of any principal amount outstanding under the Facility Agreement on each of September 26, 2011 and 2012, and 34% of such principal amount outstanding on September 26, 2013. Additionally, any amounts drawn under the Facility Agreement may become immediately due and payable upon (i) an “event of default,” as defined in the Facility Agreement, in which case the Lenders would have the right to require us to re-pay 100% of the principal amount of the loan, plus any accrued and unpaid interest thereon, or (ii) the consummation of certain change of control transactions, in which case the Lenders would have the right to require us to re-pay 110% of the outstanding principal amount of the loan, plus any accrued and unpaid interest thereon. An event of default under the Facility Agreement will occur
34
Table of Contents
if, among other things, (i) we fail to make payment when due; (ii) we fail to comply in any material respect with any covenant of the Facility Agreement, and such failure is not cured; (iii) any representation or warranty made by us in any transaction document was incorrect, false, or misleading in any material respect as of the date it was made; (iv) we are generally unable to pay our debts as they become due or a bankruptcy or similar proceeding has commenced by or against us; and (v) cash and cash equivalents on the last day of each calendar quarter are less than $10 million. The Facility Agreement also contains customary covenants regarding operations of our business. As of December 31, 2009, we are in compliance with all the covenants under the Facility Agreement.
In connection with the Facility Agreement, we entered into a security agreement with the Lenders, pursuant to which, as security for our repayment obligations under the Facility Agreement, we granted to the Lenders a security interest in certain of our intellectual property, including intellectual property relating to Xibrom, Istalol, Vitrase and each other product marketed by or under license from us, and certain personal property relating thereto.
During 2009, we generated $4.0 million of cash from operations. The positive cash flow we generated from operations was primarily the result of the growth of our business, offset by changes in operating assets and liabilities. We had a net loss of $57.8 million, offset by the change in operating assets and liabilities of $2.0 million and non-cash charges totaling $59.8 million. Non-cash charges primarily include $52.1 million in loss on warrant valuation, $3.7 million in stock-based compensation costs, $3.1 million in amortization of discount on the Facility Agreement, $1.0 million in depreciation and amortization, $1.1 million in amortization of deferred financing costs and ($1.2) million for the change in value of the derivative associated with the Facility Agreement.
During 2008, we used $22.5 million of cash for operations primarily as a result of the net loss of $34.7 million (as adjusted) offset by non-cash stock-based compensation costs of $4.0 million, amortization of deferred financing costs related to the senior subordinated convertible notes of $0.4 million (as adjusted), amortization of deferred financing costs related to the Facility Agreement of $0.2 million, change in the value of the convertible notes derivative of $0.1 million, change in the value of the Facility Agreement derivative of $0.4 million, amortization of discount on convertible notes of $2.8 million (as adjusted), amortization of discount on Facility Agreement of $0.5 million, loss on extinguishment of debt of $2.5 million, depreciation and amortization expense of $1.0 million and a change in operating assets and liabilities of $0.3 million.
During 2007, we used $32.0 million of cash for operations primarily as a result of the net loss of $41.8 million (as adjusted), offset by non-cash stock-based compensation costs of $3.5 million, amortization of deferred financing costs related to the convertible notes of $0.4 million, change in the value of the convertible notes derivative of $0.2 million, amortization of discount on convertible notes of $3.8 million (as adjusted), depreciation and amortization expense of $0.7 million and a change in operating assets and liabilities of $1.2 million.
Net cash provided by investing activities totaled $3.4 million in 2009, primarily due to the maturities of our short-term investment securities ($4.7 million), offset by the purchase of equipment ($1.3 million). Net cash provided by investing activities totaled $17.0 million during 2008, primarily attributable to the maturities of short-term investments ($15.9 million) and the reduction in restricted cash required to secure interest payments on the outstanding convertible notes ($2.4 million), offset by the purchase of equipment ($1.4 million). Net cash provided by investing activities totaled $1.4 million in 2007, primarily attributable to the maturities of short-term investments ($18.9 million) and the reduction in restricted cash required to secure interest payments on the outstanding convertible notes ($3.2 million), offset by the purchase of short-term investments ($17.9 million) and the purchase of equipment ($2.8 million).
Net cash used in financing activities totaled $2.0 million in 2009, primarily as a result of net repayments on our Revolving Credit Facility ($2.0 million). Cash provided by financing activities in 2008 totaled $28.3 million primarily the result of the issuance of debt under our Facility Agreement in the principal amount of $65.0 million, the net borrowings from the Revolving Credit Facility of $7.5 million, offset by the repayment of our convertible notes of $40.0 million and debt issuance costs of $4.3 million. Cash provided by financing activities in 2007 totaled $38.9 million primarily the result of the sale of an aggregate of 5,250,000 shares of our common stock at a purchase price of $7.00 per share, resulting in gross proceeds to us of $36.7 million before payment of offering expenses and placement agent fees of $2.7 million. Additionally, we received $3.6 million from the exercise of 1,276,116 warrants.
We believe that current cash and cash equivalents, together with amounts available for borrowing under our Revolving Credit Facility and cash generated from operations, will be sufficient to meet anticipated cash needs for operating and capital expenditures for at least the next twelve months.
35
Table of Contents
However, our actual future capital requirements will depend on many factors, including the following:
| • | the success of the commercialization of our products; |
| • | sales and marketing activities, and expansion of our commercial infrastructure, related to our approved products and product candidates; |
| • | the results of our clinical trials and requirements to conduct additional clinical trials; |
| • | the introduction of potential generic products; |
| • | the rate of progress of our research and development programs; |
| • | the time and expense necessary to obtain regulatory approvals; |
| • | activities and payments in connection with potential acquisitions of companies, products or technology; |
| • | the results of inquiries from the subpoenas received in April 2008 from the United States District Attorney’s office; |
| • | competitive, technological, market and other developments; and |
| • | our ability to establish and maintain partnering relationships. |
These factors may cause us to seek to raise additional funds through additional sales of our debt, or equity or other securities. There can be no assurance that funds from these sources will be available when needed or, if available, will be on terms favorable to us or to our stockholders. If additional funds are raised by issuing equity securities, the percentage ownership of our stockholders will be reduced, stockholders may experience additional dilution or such equity securities may provide for rights, preferences or privileges senior to those of the holders of our common stock.
We have never been profitable, and, while we currently anticipate becoming profitable in 2010, we might never become profitable. As of December 31, 2009, our accumulated deficit was $397.3 million (including a non-cash valuation warrant adjustment of $52.1 million), including a net loss of approximately $57.8 million for the year ended December 31, 2009 and a stockholders’ deficit of $78.0 million.
In April 2008 we received subpoenas from the office of the U.S. Attorney for the Western District of New York requesting information regarding the marketing activities related to Xibrom. From April 2008 through December 31, 2009, we incurred approximately $2.8 million in legal fees associated with this matter and expect to incur significant expenses in the future. In addition, if the government chooses to engage in civil litigation or initiate a criminal prosecution against us as a result of its review of the requested documents and other evidence, we may have to incur significant amounts to defend such action or pay or incur substantial fines or penalties, either of which could significantly deplete our cash resources. As of December 31, 2009 such legal costs are neither reasonably estimable nor probable.
The DoD’s TRICARE Retail Pharmacy program pursuant to section 703 of the National Defense Authorization Act of 2008, became effective on May 26, 2009. This regulation would require manufacturers to pay rebates to the DoD on products distributed to TRICARE beneficiaries through retail pharmacies retroactive to January 28, 2008. The regulation requires that pharmaceuticals paid for by the DoD through the TRICARE Retail Pharmacy program be subject to the Federal Ceiling Price program, which would require manufacturers to provide DoD with a refund on pharmaceuticals utilized through the TRICARE Retail Pharmacy program. We have requested a waiver of the retroactive rebate for TRICARE Retail Pharmacy utilization for the period from January 28, 2008 to May 26, 2009. In addition, the regulation is currently the subject of litigation, and it is our belief that the retroactive application of the regulation is contrary to established case law. We have determined that payment of the retroactive rebate created by the regulation is neither reasonably estimable nor probable as of December 31, 2009.
We incurred legal expenses in the amount of $0.5 million and $1.2 million with the law firm, Covington & Burling, during 2009 and 2008 respectively. A member of our Board of Directors is senior counsel in this law firm.
Contractual Obligations
The following table summarizes our contractual obligations as of December 31, 2009 (in thousands):
| Payments due by period | |||||||||||||||
| Total | Less than 1 year |
1-3 years |
3-5 years |
More than 5 years | |||||||||||
| Operating Lease Obligations |
$ | 2,016 | $ | 1,071 | $ | 364 | $ | 350 | $ | 231 | |||||
| Obligation Under Capital Leases |
211 | 142 | 69 | — | — | ||||||||||
| Revolving Credit Facility |
13,000 | 13,000 | — | — | — | ||||||||||
| Facility Agreement (1) |
80,844 | 4,225 | 51,350 | 25,269 | — | ||||||||||
| Other Long-term Debt Obligations (2) |
88 | 28 | 49 | 11 | — | ||||||||||
| Total: |
$ | 96,159 | $ | 18,466 | $ | 51,832 | $ | 25,630 | $ | 231 | |||||
| (1) | Includes $65.0 million in principal amount of our Facility Agreement, bearing 6.50% interest per annum payable quarterly in cash in arrears. The Facility Agreement expires September 2013. We are required to repay 33% of any principal amounts outstanding under the Facility Agreement on each of September 26, 2011 and 2012, and 34% of such principal amount outstanding on September 26, 2013. |
| (2) | In May 2006, we entered into a supply arrangement with SPS Biomedia Ltd. to supply hyaluronidase. As part of the agreement, we were obligated to fund a portion of the construction costs, up to $200,000, for the build out of the laboratory. The contract required an upfront payment of $70,000 and annual payment of $20,000 at an interest rate of 11% per annum. |
36
Table of Contents
In addition to the above, we are committed to make potential future milestone payments to third-parties as part of our in-licensing and development programs. Milestone payments under these agreements generally become due and payable only upon achievement of certain development, regulatory and/or commercial milestones. Because the achievement of these milestones is neither probable nor reasonably estimable, such contingencies have not been recorded on our consolidated balance sheet. As of December 31, 2009, the maximum potential future milestone payments to third-parties are $27.0 million.
Critical Accounting Policies and Estimates
Management’s discussion and analysis of financial condition and results of operations, as well as disclosures included elsewhere in this Annual Report on Form 10-K, are based upon our consolidated financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. Our significant accounting policies are described in the notes to the audited consolidated financial statements contained elsewhere in this Annual Report on Form 10-K. Included within these policies are our “critical accounting policies.” Critical accounting policies are those policies that are most important to the preparation of our consolidated financial statements and require management’s most subjective and complex judgment due to the need to make estimates about matters that are inherently uncertain. Although we believe that our estimates and assumptions are reasonable, actual results may differ significantly from these estimates. Changes in estimates and assumptions based upon actual results may have a material impact on our results of operations and/or financial condition.
We believe that the critical accounting policies that most impact the consolidated financial statements are as described below.
Revenue Recognition
Product Revenues. We recognize revenues from product sales when there is persuasive evidence that an arrangement exists, when title has passed, the price is fixed or determinable, and we are reasonably assured of collecting the resulting receivable. We recognize product revenues of estimated allowances for discounts, product returns, wholesaler fees, rebates and chargebacks. If actual future payments and credits for allowances for discounts, product returns, wholesaler fees, rebates and chargebacks exceed the estimates we made at the time of sale, our financial position, results of operations and cash flows would be negatively impacted.
We establish allowances for estimated rebates, chargebacks and product returns based on numerous qualitative and quantitative factors, including:
| • | the number of and specific contractual terms of agreements with customers; |
| • | estimated level of units in the distribution channel; |
| • | historical rebates, chargebacks and returns of products; |
| • | direct communication with customers; |
| • | anticipated introduction of competitive products or generics; |
| • | anticipated pricing strategy changes by us and/or our competitors; |
| • | analysis of prescription data gathered by a third-party prescription data provider; |
| • | the impact of wholesaler distribution agreements; |
| • | the impact of changes in state and federal regulations; and |
| • | the estimated remaining shelf life of products. |
In our analyses, we utilize on hand unit data purchased from the major wholesalers, as well as prescription data purchased from a third-party data provider, to develop estimates of historical unit channel pull-through. We utilize an internal analysis to compare historical net product shipments to both estimated historical prescriptions written and historical returns. Based on that analysis, we develop an estimate of the quantity of product which may be subject to various discounts, product returns, rebates, chargebacks and wholesaler fees.
37
Table of Contents
We record estimated allowances for rebates, chargebacks, product returns and other allowances in the same period when revenue is recognized. The objective of recording the allowances for such deductions at the time of sale is to provide a reasonable estimate of the aggregate amount of credit to our direct customers or payments to our indirect customers. Customers typically process their claim for allowances such as early pay discounts promptly, usually within the established payment terms. We monitor actual credit memos issued to our customers and compare such actual amounts to the estimated provisions, in the aggregate, for each allowance category to assess the reasonableness of the various reserves at each balance sheet date. Differences between our estimated allowances and actual credits issued have not been significant, and are accounted for in the current period as a change in estimate in accordance with generally accepted accounting principles.
In general, we are obligated to accept from our customers the return of pharmaceutical products that have reached their expiration date. We authorize returns for damaged products and, on a limited basis, exchanges for expiring and expired products in accordance with our return goods policy and procedures, and have established reserves for such amounts at the time of sale. We typically refund the agreed proportion of the sales price by the issuance of a credit, rather than cash refund or exchanges for inventory, and the returned product is destroyed. With the launch of each of our products, we recorded a sales return allowance, which is larger for stocking orders than subsequent re-orders. To date, actual product returns have not exceeded our estimated allowances for returns. Although we believe that our estimates and assumptions are reasonable as of the date when made, actual results may differ significantly from these estimates. Our financial position, results of operations and cash flows may be materially and negatively impacted if actual returns exceed our estimated allowances for returns.
We identify product returns by their manufacturing lot number. Because we manufacture in bulk, lot sizes can be large and, as a result, sales of any individual lot may occur over several periods. As a result, we are unable to specify if actual returns or credits relate to a sale that occurred in the current period or a prior period, and therefore, we cannot specify how much of the allowance recorded relates to sales made in prior periods. Since there have been no material differences between estimates recorded and actual credits issued, we believe our systems and procedures are adequate for managing our business.
Allowances for product returns were $5.5 and $3.2 million as of December 31, 2009 and 2008, respectively. These allowances reflect an estimate of the our liability for products that may be returned by the original purchaser in accordance with our stated return policy, which allows customers to return products within six months of their respective expiration dates and for a period up to twelve months after such products have reached their respective expiration dates. We estimate our liability for product returns at each reporting period based on the estimated units in the channel and the other factors discussed above. Our absolute exposure for product returns has increased as a result of increased sales of our existing products and our additional historical sales data upon which to analyze these allowances. Accordingly, reductions to revenue and corresponding increases to allowance accounts have likewise increased.
As a percentage of gross product revenues, the allowance for product returns was 3.7%, 3.7% and 3.3% for the years ended December 31, 2009, 2008 and 2007, respectively. We typically apply a higher allowance for product returns on stocking orders in connection with an initial launch.
We also periodically offer promotional discounts to our existing customer base. These discounts are calculated as a percentage of the current published list price. Accordingly, the discounts are recorded as a reduction of revenue in the period that the program is offered. In addition to promotional discounts, which are recorded at the time that we implement a price increase, we generally offer our existing customer base an opportunity to purchase a limited quantity of products at the previous list price. Shipments resulting from these programs generally are not in excess of ordinary levels and therefore, we recognize the related revenue upon receipt by the customer and include the sale in estimating our various product-related allowances. In the event we determine that these sales represent purchases of inventory in excess of ordinary levels for a given wholesaler, the potential impact on product returns exposure would be specifically evaluated and reflected as a reduction to revenues at the time of such sale.
Allowances for estimated rebates, chargebacks and other discounts such as wholesaler fees were $6.4 million and $2.9 million as of December 31, 2009 and 2008, respectively. These allowances reflect an estimate of our liability for items such as rebates due to various governmental organizations under the Medicare/Medicaid regulations, rebates due to managed-care organizations under specific contracts, chargebacks due to various organizations purchasing certain of our products through federal contracts and/or group purchasing agreements and fees charged by certain wholesalers under distribution agreements. We estimate our liability for rebates, chargebacks and other discounts such as wholesaler fees at each reporting period based on a combination of quantitative and qualitative assumptions listed above.
As a percentage of gross product revenues, the allowance for rebates, chargebacks and other discounts, such as wholesaler fees, was 14.7%, 9.8% and 7.5% for the years ended December 31, 2009, 2008 and 2007, respectively. The increase is due in part to the increase in the number of managed care contracts, federal contracts and wholesaler distribution agreements entered into during 2009 and 2008.
38
Table of Contents
License Revenue. Amounts received for product and technology license fees under multiple-element arrangements are deferred and recognized over the period of such services or performance if such arrangements require on-going services or performance. Amounts received for milestones are recognized upon achievement of the milestone, unless we have ongoing performance obligations. Any amounts received prior to satisfying our revenue recognition criteria will be recorded as deferred income in the accompanying consolidated balance sheets. During the year ended December 31, 2009, we recognized $3.1 million of previously deferred income primarily related to the termination of our Supply Agreement with Otsuka.
Inventory
Inventories relate to: (i) Xibrom, a topical non-steroidal anti-inflammatory formulation of bromfenac for the treatment of ocular inflammation and pain following cataract surgery; (ii) Bepreve, for the treatment of ocular itching associated with allergic conjunctivitis; (iii) Istalol, for the treatment of glaucoma; and (iv) Vitrase 200 USP units/ml for use as a spreading agent to facilitate the absorption and dispersion of other injected drugs. Inventories, net of allowances, are stated at the lower of cost or market. Cost is determined by the first-in, first-to-expire method.
Inventories are reviewed periodically for slow-moving or obsolete status. We adjust our inventory to reflect situations in which the cost of inventory is not expected to be recovered. We would record a reserve to adjust inventory to its net realizable value if: (i) a launch of a new product is delayed, inventory may not be fully utilized and could be subject to impairment, (ii) when a product is close to expiration and not expected to be sold, (iii) when a product has reached its expiration date or (iv) when a product is not expected to be saleable. In determining the reserves for these products, we consider factors such as the amount of inventory on hand and its remaining shelf life, and current and expected market conditions, including management forecasts and levels of competition. We have evaluated the current level of inventory considering historical trends and other factors, and based on our evaluation, have recorded adjustments to reflect inventory at its net realizable value. These adjustments are estimates, which could vary significantly from actual results if future economic conditions, customer demand, competition or other relevant factors differ from expectations. These estimates require us to make assessments about the future demand for our products in order to categorize the status of such inventory items as slow-moving, obsolete or in excess-of-need. These future estimates are subject to the ongoing accuracy of our forecasts of market conditions, industry trends, competition and other factors. Differences between our estimated reserves and actual inventory adjustments have not been significant, and are accounted for in the current period as a change in estimate in accordance with generally accepted accounting principles.
Stock-based Compensation
We recognize compensation costs for all stock-based awards made to employees and directors. The fair value of stock-based awards is estimated at grant date using an option pricing model and the portion that is ultimately expected to vest is recognized as compensation cost over the requisite service period.
Since stock-based compensation is recognized only for those awards that are ultimately expected to vest, we have applied an estimated forfeiture rate to unvested awards for the purpose of calculating compensation cost. These estimates will be revised, if necessary, in future periods if actual forfeitures differ from estimates. Changes in forfeiture estimates impact compensation cost in the period in which the change in estimate occurs.
We use the Black-Scholes option-pricing model to estimate the fair value of stock-based awards. The determination of fair value using the Black-Scholes option-pricing model is affected by our stock price as well as assumptions regarding a number of complex and subjective variables, including expected stock price volatility, risk-free interest rate, expected dividends and projected employee stock option exercise behaviors. We estimate the expected term based on the contractual term of the awards and employees’ exercise and expected post-vesting termination behavior.
At December 31, 2009, there was $5.2 million of total unrecognized compensation cost related to non-vested stock options, which is expected to be recognized over a remaining weighted average vesting period of approximately 2.4 years.
Income Taxes
We record a full valuation allowance to reduce our deferred tax assets to the amount that is more likely than not to be realized. While we have considered future taxable income and ongoing prudent and feasible tax planning strategies in assessing the need for the valuation allowance, in the event we were to determine that we would be able to realize our deferred tax assets in the future in excess of its net recorded amount, an adjustment to the deferred tax asset would increase income in the period such determination was made.
39
Table of Contents
Changes in Accounting Principles
Effective January 1, 2009, we changed the classification of our warrants from equity to debt as required by the Derivatives and Hedging Topic of the FASB Accounting Standards Codification. The cumulative effect was a reduction to additional paid-in capital of $10.7 million to reclassify the value of warrants from equity to a liability and a decrease in accumulated deficit of $3.7 million to reflect the change in the value of the warrants between their issuance date and January 1, 2009. The warrants will be carried at estimated fair value and adjusted quarterly through earnings.
Effective January 1, 2009, we have accounted for the liability and equity components of our convertible notes in a manner that reflects our non-convertible debt borrowing rate when interest cost is recognized as required by the Debt with Conversion and Other Options Topic of the FASB Accounting Standards Codification. Additionally, debt issuance costs are required to be allocated in proportion to the allocation of the liability and equity components and accounted for as debt issuance costs and equity issuance costs, respectively. This change required retrospective application and, accordingly, the prior periods’ financial statements included herein have been adjusted.
We determined that the fair value of the convertible notes at issuance in 2006, using a non-convertible debt borrowing rate of 21%, was approximately $28.6 million, and designated the residual value of approximately $11.4 million as the equity component, recorded as a debt discount. Additionally, we allocated approximately $2.0 million of the $2.8 million original convertible notes issuance cost as debt issuance cost and the remaining $0.8 million as equity issuance cost. Because the convertible notes were extinguished in September 2008, the balances of the liability and equity components were zero for each of the periods ended December 31, 2009 and 2008, respectively.
The remaining debt discount was amortized to interest expense over the expected life of the convertible notes using the effective interest method, applying a 21% effective interest rate. The convertible notes were issued June 22, 2006 and were due June 22, 2011.
Interest expense related to the convertible notes was recognized as follows (in thousands):
| Years Ended December 31, | |||||||||
| 2009 | 2008 (as adjusted) |
2007 (as adjusted) | |||||||
| Interest expense — stated coupon rate |
$ | — | $ | 2,367 | $ | 3,200 | |||
| Interest expense — amortization of debt discount |
— | 2,842 | 3,789 | ||||||
| Total interest expense — convertible notes |
$ | — | $ | 5,209 | $ | 6,989 | |||
40
Table of Contents
Effect of Change in Accounting Principle to Consolidated Balance Sheet
The following table summarizes the effect of the change in accounting principle related to our convertible notes on the consolidated balance sheet as of December 31, 2008 (in thousands):
| December 31, 2008 | ||||||||||||
| As Previously Reported |
As Adjusted | Effect of Change |
||||||||||
| Assets: |
||||||||||||
| Cash and cash equivalents |
$ | 48,316 | $ | 48,316 | $ | — | ||||||
| Short-term investments |
4,700 | 4,700 | — | |||||||||
| Accounts receivable, net |
15,242 | 15,242 | — | |||||||||
| Inventory, net |
2,289 | 2,289 | — | |||||||||
| Other current assets |
2,150 | 2,150 | — | |||||||||
| Total current assets |
72,697 | 72,697 | — | |||||||||
| Property and equipment, net |
5,728 | 5,728 | — | |||||||||
| Deferred financing costs, net |
4,028 | 4,028 | — | |||||||||
| Deposits and other assets |
207 | 207 | — | |||||||||
| Total assets |
$ | 82,660 | $ | 82,660 | $ | — | ||||||
| Liabilities: |
||||||||||||
| Accounts payable |
$ | 5,386 | $ | 5,386 | $ | — | ||||||
| Accrued compensation and related expenses |
3,945 | 3,945 | — | |||||||||
| Revolving Credit Facility |
15,000 | 15,000 | — | |||||||||
| Current portion of long-term liabilities |
310 | 310 | — | |||||||||
| Current portion of obligations under capital leases |
117 | 117 | — | |||||||||
| Allowance for rebates and chargebacks |
2,074 | 2,074 | — | |||||||||
| Allowance for product returns |
3,241 | 3,241 | — | |||||||||
| Royalties payable |
5,867 | 5,867 | — | |||||||||
| Other accrued expenses |
5,257 | 5,257 | — | |||||||||
| Total current liabilities |
41,197 | 41,197 | — | |||||||||
| Deferred rent |
213 | 213 | — | |||||||||
| Deferred income |
3,055 | 3,055 | — | |||||||||
| Obligations under capital leases |
187 | 187 | — | |||||||||
| Long-term liabilities |
50 | 50 | — | |||||||||
| Facility Agreement |
55,157 | 55,157 | — | |||||||||
| Total liabilities |
99,859 | 99,859 | — | |||||||||
| Stockholders’ deficit: |
||||||||||||
| Common stock |
33 | 33 | — | |||||||||
| Additional paid-in capital |
316,650 | 326,036 | 9,386 | |||||||||
| Accumulated other comprehensive loss |
(25 | ) | (25 | ) | — | |||||||
| Accumulated deficit |
(333,857 | ) | (343,243 | ) | (9,386 | ) | ||||||
| Total stockholders’ deficit |
(17,199 | ) | (17,199 | ) | — | |||||||
| Total liabilities and stockholders’ deficit |
$ | 82,660 | $ | 82,660 | $ | — | ||||||
41
Table of Contents
Effect of Change in Accounting Principle to Consolidated Statements of Operations
The following table summarizes the effect of change in accounting principle related to our convertible notes on the consolidated statements of operations for the years ended December 31, 2008 and 2007 (in thousands, except EPS):
| Year Ended December 31, 2008 |
||||||||||||
| As Previously Reported |
As Adjusted | Effect of Change |
||||||||||
| Total revenues |
$ | 83,076 | $ | 83,076 | $ | — | ||||||
| Cost of products sold |
21,947 | 21,947 | — | |||||||||
| Costs and expenses |
85,939 | 85,939 | — | |||||||||
| Loss from operations |
(24,810 | ) | (24,810 | ) | — | |||||||
| Other (expense) income: |
||||||||||||
| Interest income |
714 | 714 | — | |||||||||
| Interest expense |
(5,403 | ) | (8,100 | ) | (2,697 | ) | ||||||
| Loss on extinguishment of debt |
(1,201 | ) | (2,497 | ) | (1,296 | ) | ||||||
| Gain on derivative valuation |
26 | 26 | — | |||||||||
| Total other expense, net |
(5,864 | ) | (9,857 | ) | (3,993 | ) | ||||||
| Net loss |
$ | (30,674 | ) | $ | (34,667 | ) | (3,993 | ) | ||||
| Net loss per common share, basic and diluted |
$ | (0.93 | ) | $ | (1.05 | ) | $ | (0.12 | ) | |||
| Shares used in computing net loss per common share, basic and diluted |
33,028 | 33,028 | — | |||||||||
| Year Ended December 31, 2007 |
||||||||||||
| As Previously Reported |
As Adjusted | Effect of Change |
||||||||||
| Total revenues |
$ | 58,867 | $ | 58,867 | $ | — | ||||||
| Cost of products sold |
15,864 | 15,864 | — | |||||||||
| Costs and expenses |
79,095 | 79,095 | — | |||||||||
| Loss from operations |
(36,092 | ) | (36,092 | ) | — | |||||||
| Other (expense) income: |
||||||||||||
| Interest income |
2,141 | 2,141 | — | |||||||||
| Interest expense |
(4,073 | ) | (7,669 | ) | (3,596 | ) | ||||||
| Loss on derivative valuation |
(197 | ) | (197 | ) | — | |||||||
| Total other expense, net |
(2,129 | ) | (5,725 | ) | (3,596 | ) | ||||||
| Net loss |
$ | (38,221 | ) | $ | (41,817 | ) | (3,596 | ) | ||||
| Net loss per common share, basic and diluted |
$ | (1.29 | ) | $ | (1.41 | ) | $ | (0.12 | ) | |||
| Shares used in computing net loss per common share, basic and diluted |
29,621 | 29,621 | — | |||||||||
42
Table of Contents
Effect of Change in Accounting Principle to Consolidated Statements of Cash Flows
The following table summarizes the effect of change in accounting principle related to our convertible notes on the consolidated statements of cash flows for the years ended December 31, 2008 and 2007 (in thousands):
| Year Ended December 31, 2008 |
||||||||||||
| As Previously Reported |
As Adjusted | Effect of Change |
||||||||||
| Operating activities |
||||||||||||
| Net loss |
$ | (30,674 | ) | $ | (34,667 | ) | $ | (3,993 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Stock-based compensation costs |
3,978 | 3,978 | — | |||||||||
| Amortization of deferred financing costs |
687 | 569 | (118 | ) | ||||||||
| Amortization of discount on convertible notes |
27 | 2,842 | 2,815 | |||||||||
| Amortization of discount on Facility Agreement |
544 | 544 | — | |||||||||
| Loss on extinguishment of debt |
1,201 | 2,497 | 1,296 | |||||||||
| Change in value of derivative related to convertible notes |
42 | 42 | — | |||||||||
| Change in value of derivative related to Facility Agreement |
354 | 354 | — | |||||||||
| Depreciation and amortization |
1,046 | 1,046 | — | |||||||||
| Changes in operating assets and liabilities |
||||||||||||
| Accounts receivable, net |
(4,081 | ) | (4,081 | ) | — | |||||||
| Inventory, net |
5 | 5 | — | |||||||||
| Other current assets |
(28 | ) | (28 | ) | — | |||||||
| Accounts payable |
1,725 | 1,725 | — | |||||||||
| Accrued compensation and related expenses |
249 | 249 | — | |||||||||
| Other accrued expenses |
2,964 | 2,964 | — | |||||||||
| Other liabilities |
(241 | ) | (241 | ) | — | |||||||
| Deferred rent |
(17 | ) | (17 | ) | — | |||||||
| Deferred income |
(278 | ) | (278 | ) | — | |||||||
| Net cash used in operating activities |
(22,497 | ) | (22,497 | ) | — | |||||||
| Investing activities |
||||||||||||
| Net cash provided by investing activities |
16,984 | 16,984 | — | |||||||||
| Financing activities |
||||||||||||
| Net cash provided by financing activities |
28,329 | 28,329 | — | |||||||||
| Increase in cash and cash equivalents |
22,816 | 22,816 | ||||||||||
| Cash and cash equivalents at beginning of year |
25,500 | 25,500 | — | |||||||||
| Cash and cash equivalents at end of year |
$ | 48,316 | $ | 48,316 | $ | — | ||||||
43
Table of Contents
| Year Ended December 31, 2007 |
||||||||||||
| As Previously Reported |
As Adjusted | Effect of Change |
||||||||||
| Operating activities |
||||||||||||
| Net loss |
$ | (38,221 | ) | $ | (41,817 | ) | $ | (3,596 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Stock-based compensation costs |
3,488 | 3,488 | — | |||||||||
| Amortization of deferred financing costs |
558 | 401 | (157 | ) | ||||||||
| Amortization of discount on convertible notes |
56 | 3,809 | 3,753 | |||||||||
| Change in value of derivative related to convertible notes |
197 | 197 | — | |||||||||
| Depreciation and amortization |
725 | 725 | — | |||||||||
| Changes in operating assets and liabilities |
||||||||||||
| Accounts receivable, net |
(4,822 | ) | (4,822 | ) | — | |||||||
| Inventory, net |
(820 | ) | (820 | ) | — | |||||||
| Other current assets |
(450 | ) | (450 | ) | — | |||||||
| Accounts payable |
1,443 | 1,443 | — | |||||||||
| Accrued compensation and related expenses |
1,385 | 1,385 | — | |||||||||
| Other accrued expenses |
5,012 | 5,012 | — | |||||||||
| Other liabilities |
(285 | ) | (285 | ) | — | |||||||
| Deferred rent |
14 | 14 | — | |||||||||
| Deferred income |
(278 | ) | (278 | ) | — | |||||||
| Net cash used in operating activities |
(31,998 | ) | (31,998 | ) | — | |||||||
| Investing activities |
||||||||||||
| Net cash provided by investing activities |
1,378 | 1,378 | — | |||||||||
| Financing activities |
||||||||||||
| Net cash provided by financing activities |
38,884 | 38,884 | — | |||||||||
| Increase in cash and cash equivalents |
8,264 | 8,264 | ||||||||||
| Cash and cash equivalents at beginning of year |
17,236 | 17,236 | — | |||||||||
| Cash and cash equivalents at end of year |
$ | 25,500 | $ | 25,500 | $ | — | ||||||
New Accounting Pronouncements
In May 2009, the FASB issued Accounting Standards Codification 855-10, “Subsequent Events”, or ASC 855-10, which establishes general standards for accounting and disclosure of events that occur after the balance sheet date but before the financial statements are issued or are available to be issued. The pronouncement requires the disclosure of the date through which an entity has evaluated subsequent events and the basis for that date, whether that date represents the date the financial statements were issued or were available to be issued. We evaluated subsequent events through the issuance date of the financial statements, which did not have a material impact on our consolidated financial statements.
In June 2009, the FASB issued Accounting Standards Codification 105-10, “Accounting Standards Codification and the Hierarchy of Generally Accepted Accounting Principles”, or ASC 105-10. ASC 105-10 became the source of authoritative U.S. Generally Accepted Accounting Principles, or GAAP, recognized by the FASB to be applied by non-government entities. It also modified the GAAP hierarchy to include only two levels of GAAP; authoritative and non-authoritative. We adopted ASC 105-10 for the reporting in 2009. The adoption did not have a material impact on our consolidated financial statements.
In August 2009, the FASB issued Accounting Standards Update No. 2009-05, Measuring Liabilities at Fair Value, or ASU 2009-05, which amends ASC 820 to provide clarification of a circumstance in which a quoted price in an active market for an identical liability is not available. A reporting entity is required to measure fair value using one or more of the following methods: 1) a valuation technique that uses a) the quoted price of the identical liability when traded as an asset or b) quoted prices for similar liabilities (or similar liabilities when traded as assets) and/or 2) a valuation technique that is consistent with the principles of ASC 820. ASU 2009-05 also clarifies that when estimating the fair value of a liability, a reporting entity is not required to adjust to include inputs relating to the existence of transfer restrictions on that liability. The adoption did not have a material impact on our consolidated financial statements.
44
Table of Contents
| Item 7A: | Quantitative and Qualitative Disclosures About Market Risk. |
The primary objective of our investment policy is to preserve principal while at the same time maximizing the income we receive from our investments without significantly increasing risk. Some of the securities that we have invested in had market risk. This means that a change in prevailing interest rates may cause the principal amount of the investment to fluctuate. For example, if we hold a security that was issued with a fixed interest rate at the then-prevailing rate and the prevailing interest rate later increases, the principal amount of our investment will probably decline. Seeking to minimize this risk, historically we maintained our portfolio of cash equivalents and short-term investments in a variety of securities, including commercial paper, money market funds, government and non-government debt securities and auction rate securities. All of our cash is held in non-interest bearing accounts, which is fully insured by the Federal Deposit Insurance Corporation. When our cash is invested in short-term investments, the average duration is usually less than one year.
All outstanding amounts under our Revolving Credit Facility bear interest at a variable rate equal to the lender’s prime rate plus a margin of 0.25%. In no event shall the interest rate on outstanding borrowings be less than 4.25%. Interest is payable on a monthly basis and may expose us to market risk due to changes in interest rates. As of December 31, 2009, we had $13.0 million outstanding under our Revolving Credit Facility. The interest rate at December 31, 2009 was 4.25%. A 10% change in interest rates on our Revolving Credit Facility would not have had a material effect on our net loss for the year ended December 31, 2009.
We have operated primarily in the United States. Accordingly, we have not had any significant exposure to foreign currency rate fluctuations.
| Item 8: | Financial Statements and Supplementary Data. |
The consolidated financial statements and supplementary data required by this item are set forth on the pages indicated in Item 15(a).
45
Table of Contents
| Item 9: | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure. |
On February 20, 2009, our audit committee approved the dismissal of Ernst & Young LLP, or E&Y, as our independent registered public accounting firm.
The audit report of E&Y on our financial statements as of and for each of the two fiscal years ended December 31, 2008 and 2007 did not contain any adverse opinion or disclaimer of opinion, nor was it qualified or modified as to uncertainty, audit scope, or accounting principles.
In connection with the audit of our financial statements for each of the two fiscal years ended December 31, 2008 and 2007, and in the subsequent interim period through February 20, 2009, the date of the dismissal of E&Y, (i) there were no disagreements with E&Y on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedures, which disagreements, if not resolved to E&Y’s satisfaction, would have caused E&Y to make reference to the subject matter of the disagreement in connection with its report, and (ii) there were no “reportable events,” as that term is described in Item 304(a)(1)(v) of Regulation S-K.
E&Y has provided us with a letter stating that they agree that there were no disagreements during fiscal years ended December 31, 2008 and 2007, and through February 20, 2009, and we filed a copy of such letter as Exhibit 16.1 to our Current Report on Form 8-K, filed on February 26, 2009, which was within the time periods prescribed by the SEC.
On March 6, 2009, our audit committee approved the appointment and engagement of BDO Seidman, LLP, or BDO, to serve as our independent registered public accounting firm, effective as of March 6, 2009.
During the Company’s two most recent fiscal years and in the subsequent interim period through March 6, 2009, neither the Company, nor anyone acting on its behalf, consulted with BDO regarding either: (i) the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit opinion that might be rendered on our financial statements, and no written report nor oral advice was provided by BDO, or (ii) any matter that was either the subject of a disagreement, as that term is defined in Item 304(a)(1)(iv) of Regulation S-K, or a reportable event, as that term is defined in Item 304(a)(1)(v) of Regulation S-K.
| Item 9A: | Controls and Procedures. |
(a) Evaluation of Disclosure Controls and Procedures
Our management, with the participation and under the supervision of our Chief Executive Officer and Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13(a) – 15(e) and 15(d) – 15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act) as of the end of the period covered by this Annual Report. The Chief Executive Officer and Chief Financial Officer have concluded, based on their evaluation of these controls and procedures, that our disclosure controls and procedures were effective as of the end of the period covered by this Annual Report to provide reasonable assurance that information required to be disclosed by us in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified in applicable SEC rules and forms. A controls system, no matter how well designed and operated, cannot provide absolute assurance that the objectives of the controls are met, and no evaluation of controls can provide absolute assurance that all controls and instances of fraud, if any, within a company have been detected.
(b) Changes in Internal Control over Financial Reporting and Remediation Plans
We have not made any significant changes to our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) during the three-month period ended December 31, 2009 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
46
Table of Contents
(c) Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, our principal executive and principal financial officers and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
| • | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; |
| • | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and |
| • | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2009. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO, in Internal Control-Integrated Framework.
Based on our assessment, management believes that, as of December 31, 2009, our internal control over financial reporting is effective based on those criteria.
Our independent registered public accounting firm has issued a report on our assessment of our internal control over financial reporting. This report appears below.
There was no change in our internal control over financial reporting that occurred during our most recently completed fiscal quarter that materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
47
Table of Contents
(d) Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of ISTA Pharmaceuticals, Inc.
We have audited ISTA Pharmaceuticals, Inc.’s (the “Company”) internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the “COSO criteria”). ISTA Pharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the effectiveness of the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, ISTA Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2009, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheet of ISTA Pharmaceuticals, Inc. as of December 31, 2009, and the related consolidated statements of operations, stockholders’ deficit, and cash flows for the year ended December 31, 2009 of ISTA Pharmaceuticals, Inc. and our report dated February 24, 2010 expressed an unqualified opinion thereon.
| /s/ BDO Seidman LLP |
Orange County, California
February 24, 2010
48
Table of Contents
| Item 9B: | Other Information. |
None.
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance |
In accordance with Instruction G (3) to Form 10-K, the information required by this Item will be provided in an amendment to this Form 10-K to be filed not later than 120 days after the end of the fiscal year covered by this Form 10-K.
| Item 11. | Executive Compensation |
In accordance with Instruction G (3) to Form 10-K, the information required by this Item will be provided in an amendment to this Form 10-K to be filed not later than 120 days after the end of the fiscal year covered by this Form 10-K.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
In accordance with Instruction G (3) to Form 10-K, the information required by this Item will be provided in an amendment to this Form 10-K to be filed not later than 120 days after the end of the fiscal year covered by this Form 10-K, with the exception of the information regarding securities authorized for issuance under our equity compensation plans, which is set forth in Item 5 of this Annual Report on Form 10-K under the heading “Equity Compensation Plans” and is incorporated herein by reference.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
In accordance with Instruction G (3) to Form 10-K, the information required by this Item will be provided in an amendment to this Form 10-K to be filed not later than 120 days after the end of the fiscal year covered by this Form 10-K.
| Item 14. | Principal Accounting Fees and Services |
In accordance with Instruction G (3) to Form 10-K, the information required by this Item will be provided in an amendment to this Form 10-K to be filed not later than 120 days after the end of the fiscal year covered by this Form 10-K.
Consistent with Section 10A (i) (2) of the Securities Exchange Act of 1934, as added by Section 202 of the Sarbanes-Oxley Act of 2002, we are responsible for listing the non-audit services approved by our Audit Committee to be performed by BDO Seidman LLP, our external auditor. Non-audit services are defined as services other than those provided in connection with an audit or a review of our financial statements. The Audit Committee has approved BDO Seidman LLP for non-audit services related to the preparation of federal and state income tax returns, and tax advice in preparing for and in connection with such filings.
49
Table of Contents
PART IV
| Item 15: | Exhibits and Financial Statement Schedules. |
(a) Financial Statements
(1) Index to Consolidated Financial Statements
The financial statements required by this item are submitted in a separate section beginning on page F-1 of this report.
CONSOLIDATED FINANCIAL STATEMENTS OF ISTA PHARMACEUTICALS, INC.
| (2) Financial Statement Schedules |
| S-1 | ||
| This financial statement schedule should be read in conjunction with the consolidated financial statements. Financial statement schedules not included in this Annual Report on Form 10-K have been omitted because they are not applicable or the required information is shown in the financial statements or notes thereto. |
||
| (3) Exhibits |
See Exhibit Index
50
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form 10-K and has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Irvine, State of California, on February 24, 2010.
| By: | /S/ VICENTE ANIDO, JR., PH.D. | |
| Vicente Anido, Jr., Ph.D. | ||
| President and Chief Executive Officer |
POWER OF ATTORNEY
Each person whose signature appears below constitutes and appoints each of Vicente Anido, Jr., Ph.D. and Lauren P. Silvernail as his or her attorney-in-fact, with full power of substitution, for him or her in any and all capacities, to sign any amendments to this Form 10-K, and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each attorney-in-fact, or his substitute, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Form 10-K has been signed by the following persons in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /S/ VICENTE ANIDO, JR., PH.D. Vicente Anido, Jr., Ph.D. |
President, Chief Executive Officer and Director | February 24, 2010 | ||
| /S/ LAUREN P. SILVERNAIL Lauren P. Silvernail |
Chief Financial Officer, Chief Accounting Officer and Vice President, Corporate Development | February 24, 2010 | ||
| /S/RICHARD C. WILLIAMS Richard C. Williams |
Director (Chairman of the Board of Directors) | February 24, 2010 | ||
| /S/ PETER BARTON HUTT Peter Barton Hutt |
Director | February 24, 2010 | ||
| /S/ KATHLEEN D. LAPORTE Kathleen D. LaPorte |
Director | February 24, 2010 | ||
| /S/ BENJAMIN F. MCGRAW III Benjamin F. McGraw III |
Director | February 24, 2010 | ||
| /S/ DEAN J. MITCHELL Dean J. Mitchell |
Director | February 24, 2010 | ||
| /S/ ANDREW J. PERLMAN Andrew J. Perlman |
Director | February 24, 2010 | ||
| /S/ WAYNE I. ROE Wayne I. Roe |
Director | February 24, 2010 | ||
51
Table of Contents
ISTA PHARMACEUTICALS, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of ISTA Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheet of ISTA Pharmaceuticals, Inc. as of December 31, 2009, and the related consolidated statements of operations, stockholders’ deficit, and cash flows for the year ended December 31, 2009. We have also audited the 2009 information included in the schedule listed in the accompanying index. These financial statements and schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and schedule based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements and schedule are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements and schedule. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement and schedule presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of ISTA Pharmaceuticals, Inc. at December 31, 2009, and the consolidated results of its operations and its cash flows for the year ended December 31, 2009, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the 2009 information in the schedule presents fairly, in all material respects, the 2009 information set forth herein.
Effective January 1, 2009, the Company changed its accounting principles related to the classification of warrants required by the Derivatives and Hedging Topic of the FASB Accounting Standards Codification and its accounting for the liability and equity components of their convertible notes required by the Debt with Conversion and Other Options Topic of the FASB Accounting Standards Codification.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), ISTA Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 24, 2010 expressed an unqualified opinion thereon.
/s/ BDO Seidman LLP
Orange County, California
February 24, 2010
F-2
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of ISTA Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheet of ISTA Pharmaceuticals, Inc. as of December 31, 2008, and the related consolidated statements of operations, stockholders’ deficit, and cash flows for each of the two years in the period ended December 31, 2008. Our audits also included the financial statement schedule listed in the Index at Item 15(a) 2. These financial statements and schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of ISTA Pharmaceuticals, Inc. at December 31, 2008, and the consolidated results of its operations and its cash flows for each of the two years in the period ended December 31, 2008, in conformity with U.S. generally accepted accounting principles. Also, in our opinion the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
As discussed in Note 1 of the consolidated financial statements, effective January 1, 2009, the Company changed its method of accounting for its convertible notes with the adoption of the guidance originally issued in FASB Staff Position APB 14-1, Accounting for Convertible Debt Instruments That May Be Settled In Cash upon Conversion (Including Partial Cash Settlement) codified in FASB ASC Topic 470-20, Debt - Debt with Conversion and Other Options.
/s/ Ernst & Young LLP
Orange County, California
February 17, 2009,
except for Note 1, as to which the date is
February 24, 2010
F-3
Table of Contents
CONSOLIDATED BALANCE SHEETS
(in thousands, except share data)
| December 31, | ||||||||
| 2009 | 2008 (as adjusted) |
|||||||
| ASSETS | ||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 53,702 | $ | 48,316 | ||||
| Short-term investments |
— | 4,700 | ||||||
| Accounts receivable, net of allowances of $94 in 2009 and $134 in 2008 |
17,434 | 15,242 | ||||||
| Inventory, net of allowance of $896 in 2009 and $612 in 2008 |
5,548 | 2,289 | ||||||
| Other current assets |
3,175 | 2,150 | ||||||
| Total current assets |
79,859 | 72,697 | ||||||
| Property and equipment, net |
6,116 | 5,728 | ||||||
| Deferred financing costs, net |
2,957 | 4,028 | ||||||
| Deposits and other assets |
212 | 207 | ||||||
| Total assets |
$ | 89,144 | $ | 82,660 | ||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 5,852 | $ | 5,386 | ||||
| Accrued compensation and related expenses |
7,731 | 3,945 | ||||||
| Revolving Credit Facility |
13,000 | 15,000 | ||||||
| Current portion of long-term liabilities |
34 | 310 | ||||||
| Current portion of obligations under capital leases |
154 | 117 | ||||||
| Allowance for rebates and chargebacks |
4,779 | 2,074 | ||||||
| Allowance for product returns |
5,509 | 3,241 | ||||||
| Royalties payable |
7,348 | 5,867 | ||||||
| Other accrued expenses |
6,339 | 5,257 | ||||||
| Total current liabilities |
50,746 | 41,197 | ||||||
| Deferred rent |
166 | 213 | ||||||
| Deferred income |
— | 3,055 | ||||||
| Obligations under capital leases |
129 | 187 | ||||||
| Long-term liabilities |
30 | 50 | ||||||
| Facility Agreement, net |
57,438 | 55,157 | ||||||
| Warrant Liability |
58,663 | — | ||||||
| Total liabilities |
167,172 | 99,859 | ||||||
| Commitments and Contingencies |
||||||||
| Stockholders’ deficit: |
||||||||
| Preferred stock, $0.001 par value; 5,000,000 shares authorized of which 1,000,000 shares have been designated as Series A Participating Preferred Stock at December 31, 2009 and 2008; no shares issued and outstanding |
— | — | ||||||
| Common stock, $0.001 par value; 100,000,000 shares authorized at December 31, 2009 and 2008; 33,291,123 and 33,079,277 shares issued and outstanding at December 31, 2009 and 2008, respectively |
33 | 33 | ||||||
| Additional paid-in capital |
319,211 | 326,036 | ||||||
| Accumulated other comprehensive loss |
— | (25 | ) | |||||
| Accumulated deficit |
(397,272 | ) | (343,243 | ) | ||||
| Total stockholders’ deficit |
(78,028 | ) | (17,199 | ) | ||||
| Total liabilities and stockholders’ deficit |
$ | 89,144 | $ | 82,660 | ||||
See accompanying notes.
F-4
Table of Contents
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share data)
| Years Ended December 31, | ||||||||||||
| 2009 | 2008 (as adjusted) |
2007 (as adjusted) |
||||||||||
| Revenues |
||||||||||||
| Product sales, net |
$ | 107,593 | $ | 82,798 | $ | 58,589 | ||||||
| License revenue |
3,055 | 278 | 278 | |||||||||
| Total revenues |
110,648 | 83,076 | 58,867 | |||||||||
| Cost of products sold |
27,278 | 21,947 | 15,864 | |||||||||
| Gross profit margin |
83,370 | 61,129 | 43,003 | |||||||||
| Costs and expenses: |
||||||||||||
| Research and development |
24,904 | 32,400 | 32,492 | |||||||||
| Selling, general and administrative |
56,377 | 53,539 | 46,603 | |||||||||
| Total costs and expenses |
81,281 | 85,939 | 79,095 | |||||||||
| Income (loss) from operations |
2,089 | (24,810 | ) | (36,092 | ) | |||||||
| Other (expense) income: |
||||||||||||
| Interest income |
— | 714 | 2,141 | |||||||||
| Interest expense |
(8,591 | ) | (8,100 | ) | (7,669 | ) | ||||||
| Loss on extinguishment of debt |
— | (2,497 | ) | — | ||||||||
| Gain (loss) on derivative valuation |
1,177 | 26 | (197 | ) | ||||||||
| Loss on warrant valuation |
(52,066 | ) | — | — | ||||||||
| Other, net |
(363 | ) | — | — | ||||||||
| Total other expense |
(59,843 | ) | (9,857 | ) | (5,725 | ) | ||||||
| Net loss |
$ | (57,754 | ) | $ | (34,667 | ) | $ | (41,817 | ) | |||
| Net loss per common share, basic and diluted |
$ | (1.74 | ) | $ | (1.05 | ) | $ | (1.41 | ) | |||
| Shares used in computing net loss per common share, basic and diluted |
33,228 | 33,028 | 29,621 | |||||||||
See accompanying notes.
F-5
Table of Contents
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ DEFICIT
(in thousands, except share data)
| Common Stock | Additional Paid-in |
Accumulated Other Comprehensive |
Accumulated | Total Stockholders’ Equity |
|||||||||||||||||
| Shares | Amount | Capital | Loss | Deficit | (Deficit) | ||||||||||||||||
| Balance at December 31, 2006, as previously reported |
26,193,745 | $ | 26 | $ | 260,401 | $ | (24 | ) | $ | (264,962 | ) | $ | (4,559 | ) | |||||||
| Effect of change in accounting principle |
— | — | 10,582 | — | (1,797 | ) | 8,785 | ||||||||||||||
| Balance at December 31, 2006 (as adjusted) |
26,193,745 | 26 | 270,983 | (24 | ) | (266,759 | ) | 4,226 | |||||||||||||
| Issuance of common stock for options |
33,763 | 1 | 195 | — | — | 196 | |||||||||||||||
| Restricted stock issuances |
139,927 | — | — | — | — | — | |||||||||||||||
| Common stock issued under ESPP |
18,336 | — | 112 | — | — | 112 | |||||||||||||||
| Issuance of common stock in conjunction with the registered direct public offerings, net of issuance costs of $2,677 |
5,250,000 | 5 | 34,048 | — | — | 34,053 | |||||||||||||||
| Warrant exercise |
1,276,116 | 1 | 3,613 | — | — | 3,614 | |||||||||||||||
| Stock-based compensation costs |
— | — | 3,488 | — | — | 3,488 | |||||||||||||||
| Net loss (as adjusted) |
— | — | — | — | (41,817 | ) | (41,817 | ) | |||||||||||||
| Foreign currency translation adjustment |
— | — | — | (1 | ) | — | (1 | ) | |||||||||||||
| Unrealized gain on investments |
— | — | — | 10 | — | 10 | |||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (41,808 | ) | ||||||||||||||
| Balance at December 31, 2007(as adjusted) |
32,911,887 | 33 | 312,439 | (15 | ) | (308,576 | ) | 3,881 | |||||||||||||
| Issuance of common stock for options |
1,666 | — | 6 | — | — | 6 | |||||||||||||||
| Restricted stock issuances |
142,936 | — | — | — | — | — | |||||||||||||||
| Common stock issued under ESPP |
22,788 | — | 68 | — | — | 68 | |||||||||||||||
| Warrant issuance |
— | — | 10,741 | — | — | 10,741 | |||||||||||||||
| Stock-based compensation costs |
— | — | 3,978 | — | — | 3,978 | |||||||||||||||
| Extinguishment of conversion option on convertible notes |
— | — | (1,196 | ) | — | — | (1,196 | ) | |||||||||||||
| Net loss (as adjusted) |
— | — | — | — | (34,667 | ) | (34,667 | ) | |||||||||||||
| Unrealized loss on investments |
— | — | — | (10 | ) | — | (10 | ) | |||||||||||||
| Comprehensive loss |
— | — | — | — | — | (34,677 | ) | ||||||||||||||
| Balance at December 31, 2008 (as adjusted) |
33,079,277 | 33 | 326,036 | (25 | ) | (343,243 | ) | (17,199 | ) | ||||||||||||
| Issuance of common stock for options |
52,425 | — | 166 | — | — | 166 | |||||||||||||||
| Restricted stock issuances |
139,213 | — | — | — | — | — | |||||||||||||||
| Common stock issued under ESPP |
20,208 | — | 12 | — | — | 12 | |||||||||||||||
| Warrant reclassification to liability pursuant to ASC 815-40 |
— | — | (10,741 | ) | — | 3,725 | (7,016 | ) | |||||||||||||
| Stock-based compensation costs |
— | — | 3,738 | — | — | 3,738 | |||||||||||||||
| Net loss |
— | — | — | — | (57,754 | ) | (57,754 | ) | |||||||||||||
| Foreign currency translation adjustment |
— | — | — | 25 | — | 25 | |||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (57,729 | ) | ||||||||||||||
| Balance at December 31, 2009 |
33,291,123 | $ | 33 | $ | 319,211 | $ | — | $ | (397,272 | ) | $ | (78,028 | ) | ||||||||
See accompanying notes.
F-6
Table of Contents
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| Years Ended December 31, | ||||||||||||
| 2009 | 2008 (as adjusted) |
2007 (as adjusted) |
||||||||||
| OPERATING ACTIVITIES |
||||||||||||
| Net loss |
$ | (57,754 | ) | $ | (34,667 | ) | $ | (41,817 | ) | |||
| Adjustments to reconcile net loss to net cash provided by (used in) operating activities: |
||||||||||||
| Stock-based compensation costs |
3,738 | 3,978 | 3,488 | |||||||||
| Amortization of deferred financing costs |
1,115 | 569 | 401 | |||||||||
| Amortization of discount on convertible notes |
— | 2,842 | 3,809 | |||||||||
| Amortization of discount on Facility Agreement |
3,038 | 544 | — | |||||||||
| Loss on extinguishment of debt |
— | 2,497 | — | |||||||||
| Change in value of derivatives related to convertible notes |
— | 42 | 197 | |||||||||
| Change in value of derivatives related to Facility Agreement |
(1,177 | ) | 354 | — | ||||||||
| Change in value of warrants related to Facility Agreement |
52,066 | — | — | |||||||||
| Depreciation and amortization |
1,047 | 1,046 | 725 | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accounts receivable, net |
(2,192 | ) | (4,081 | ) | (4,822 | ) | ||||||
| Inventory, net |
(3,259 | ) | 5 | (820 | ) | |||||||
| Other current assets |
(1,025 | ) | (28 | ) | (450 | ) | ||||||
| Accounts payable |
466 | 1,725 | 1,443 | |||||||||
| Accrued compensation and related expenses |
3,786 | 249 | 1,385 | |||||||||
| Other accrued expenses |
7,536 | 2,964 | 5,012 | |||||||||
| Other liabilities |
(296 | ) | (241 | ) | (285 | ) | ||||||
| Deferred rent |
(47 | ) | (17 | ) | 14 | |||||||
| Deferred income |
(3,055 | ) | (278 | ) | (278 | ) | ||||||
| Net cash provided by (used in) operating activities |
3,987 | (22,497 | ) | (31,998 | ) | |||||||
| INVESTING ACTIVITIES |
||||||||||||
| Purchases of marketable securities |
— | — | (17,873 | ) | ||||||||
| Maturities of marketable securities |
4,700 | 15,929 | 18,940 | |||||||||
| Purchases of equipment |
(1,317 | ) | (1,385 | ) | (2,769 | ) | ||||||
| Restricted cash |
— | 2,400 | 3,200 | |||||||||
| Deposits and other assets |
(5 | ) | 40 | (120 | ) | |||||||
| Net cash provided by investing activities |
3,378 | 16,984 | 1,378 | |||||||||
| FINANCING ACTIVITIES |
||||||||||||
| Proceeds from exercise of stock options |
166 | 6 | 195 | |||||||||
| Obligations under capital leases |
(139 | ) | 29 | (51 | ) | |||||||
| Proceeds from Revolving Credit Facility |
52,000 | 37,000 | 27,500 | |||||||||
| Repayments on Revolving Credit Facility |
(54,000 | ) | (29,500 | ) | (26,500 | ) | ||||||
| Proceeds from issuance of common stock, net of issuance costs |
12 | 68 | 37,780 | |||||||||
| Repayment of convertible notes |
— | (40,000 | ) | — | ||||||||
| Proceeds from issuance of Facility Agreement |
— | 65,000 | — | |||||||||
| Financing costs on issuance of convertible notes |
— | — | (40 | ) | ||||||||
| Financing costs on issuance of Facility Agreement |
(43 | ) | (4,274 | ) | — | |||||||
| Net cash (used in) provided by financing activities |
(2,004 | ) | 28,329 | 38,884 | ||||||||
| Effect of exchange rate changes on cash |
25 | — | — | |||||||||
| Increase in cash and cash equivalents |
5,386 | 22,816 | 8,264 | |||||||||
| Cash and cash equivalents at beginning of year |
48,316 | 25,500 | 17,236 | |||||||||
| Cash and cash equivalents at end of year |
$ | 53,702 | $ | 48,316 | $ | 25,500 | ||||||
| SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION: |
||||||||||||
| Cash paid during the year for interest |
$ | 4,307 | $ | 3,283 | $ | 3,247 | ||||||
| Equipment additions under capital leases |
$ | 118 | $ | 137 | $ | 229 | ||||||
See accompanying notes.
F-7
Table of Contents
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Organization and Summary of Significant Accounting Policies
The Company
ISTA Pharmaceuticals, Inc. (“ISTA” or the “Company”) was incorporated in the state of California on February 13, 1992 to discover, develop and market new remedies for diseases and conditions of the eye. We reincorporated in Delaware on August 4, 2000. Xibrom (bromfenac sodium ophthalmic solution)®, Xibrom®, XiDay™, Istalol®, Vitrase®, Bepreve™, T-Pred™, ISTA®, ISTA Pharmaceuticals, Inc.® and the ISTA logo are our trademarks, either owned or under license.
We are a commercial stage, multi-specialty pharmaceutical company developing, marketing and selling our own products in the United States. We manufacture our products through third party contracts and we in-license or acquire new technology to add to our internal development efforts from time to time.
Our products and product candidates seek to treat serious diseases of the eye and allergies and include therapies for ocular inflammation and pain, glaucoma, dry eye and ocular and nasal allergies. The U.S. prescription markets our therapies seek to address include key segments of the $5.5 billion ophthalmic pharmaceutical market and the $2.2 billion nasal allergy market.
We currently have four products available for sale in the United States and Puerto Rico: Xibrom (bromfenac sodium ophthalmic solution) for the treatment of inflammation and pain following cataract surgery, Bepreve (bepotastine besilate ophthalmic solution) for the treatment of ocular itching associated with allergic conjunctivitis, Istalol (timolol maleate ophthalmic solution) for the treatment of glaucoma, and Vitrase (hyaluronidase for injection) for use as a spreading agent.
Basis of Presentation
We have incurred losses since inception and had an accumulated deficit of $397.3 million (including a non-cash valuation warrant adjustment of $52.1 million) through December 31, 2009. Our ability to attain profitable operations is dependent upon maintaining or obtaining sufficient working capital to complete the successful development of our products, FDA approval of our products, achieving market acceptance of such products and achievement of sufficient levels of revenues to support our cost structure. We believe that our existing capital resources will enable us to fund operations for at least the next twelve months.
The consolidated financial statements include the accounts of the Company and those of its wholly owned subsidiary, Visionex Pte. Ltd. This subsidiary, which was dissolved in January 2009, had no further operations in 2009. All significant intercompany amounts have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires us to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ significantly from those estimates.
Reclassifications
Certain comparative prior year amounts in the Consolidated Financial Statements and accompanying notes have been reclassified to conform to the current year presentation. These reclassifications had no effect on previously reported operating expenses or net loss.
Fair Value of Financial Instruments
Our financial instruments include cash and cash equivalents, short-term investments, accounts receivable, accounts payable, accrued liabilities, current and long-term debt, certain derivatives related to our debt obligations and common stock warrants issued to lenders. The carrying amount of cash and cash equivalents, short-term investments, accounts receivable, accounts payable, and accrued liabilities are considered to be representative of their respective fair values because of the short-term nature of those instruments. The carrying amount of our Revolving Credit Facility approximates fair value since the interest rate approximates the market rate for debt securities with similar terms and risk characteristics. Although our Facility Agreement is considered a financial instrument, we are unable to reasonably determine fair value.
F-8
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Cash and Cash Equivalents
Cash and cash equivalents consist of cash in banks and short-term investments with maturities of three months or less when purchased. Cash and cash equivalents are carried at cost, which we believe approximates fair value because of the short-term maturity of these instruments.
Short-term Investments
Marketable securities with a maturity of more than three months from the date of purchase are considered short-term investments. During 2008, all investments were liquidated upon maturity and converted to cash except for approximately $4.7 million (par value) in auction rate securities. During January 2009, UBS AG purchased $4.7 million in auction rate securities from us. All investments are carried at fair value, with unrealized gains and losses on available-for-sale securities included as a separate component of stockholders’ deficit and unrealized gains and losses on trading securities included in earnings. The basis for computing realized gain or losses is by specific identification.
Concentration of Credit Risk
Financial instruments that potentially subject us to a significant concentration of credit risk principally consist of cash and cash equivalents, short-term investments and trade receivables. Wholesale distributors account for a substantial portion of trade receivables. At December 31, 2009, annual net revenues from AmeriSource Bergen Corp., McKesson HBOC and Cardinal Health, Inc. accounted for 17%, 40% and 35%, respectively, of our net revenues. We maintain reserves for bad debt and such losses, in the aggregate, have not exceeded our estimates.
Inventory
Inventories, net of allowances, are stated at the lower of cost or market. Cost is determined by the first-in, first-to-expire method.
Inventories are reviewed periodically for slow-moving or obsolete status. We adjust our inventory to reflect situations in which the cost of inventory is not expected to be recovered. We would record a reserve to adjust inventory to its net realizable value if: (i) a launch of a new product is delayed, inventory may not be fully utilized and could be subject to impairment, (ii) when a product is close to expiration and not expected to be sold, (iii) when a product has reached its expiration date or (iv) when a product is not expected to be saleable. In determining the reserves for these products, we consider factors such as the amount of inventory on hand and its remaining shelf life, and current and expected market conditions, including management forecasts and levels of competition. We have evaluated the current level of inventory considering historical trends and other factors, and based on our evaluation, have recorded adjustments to reflect inventory at its net realizable value. These adjustments are estimates, which could vary significantly from actual results if future economic conditions, customer demand, competition or other relevant factors differ from expectations. These estimates require us to make assessments about the future demand for our products in order to categorize the status of such inventory items as slow-moving, obsolete or in excess-of-need. These future estimates are subject to the ongoing accuracy of our forecasts of market conditions, industry trends, competition and other factors. Differences between our estimated reserves and actual inventory adjustments have not been significant, and are accounted for in the current period as a change in estimate in accordance with generally accepted accounting principles.
Costs incurred for the manufacture of validation batches for pre-approval products are recorded as research and development expenses in the period in which those costs are incurred.
Property and Equipment
Property and equipment are recorded at cost. Equipment and furniture are depreciated using the straight-line method over their estimated useful lives (generally three to seven years) and leasehold improvements are amortized using the straight-line method over the estimated useful life of the asset or the lease term, whichever is shorter. Equipment acquired under capital leases is amortized over the estimated useful life of the assets and included in depreciation expense.
Long-lived Assets
If indicators of impairment exist, we assess the recoverability of the affected long-lived assets by determining whether the carrying value of such assets can be recovered through undiscounted future operating cash flows. If impairment is indicated, we measure the amount of such impairment by comparing the fair value to the carrying value. We believe the future cash flows to be received from the long-lived assets will exceed the assets’ carrying value, and accordingly, we have not recognized any impairment losses through December 31, 2009.
F-9
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Deferred Financing Costs
In connection with the issuance of our debt agreements, including the convertible notes and the Facility Agreement, we paid financing costs, which consisted primarily of placement agent fees, accounting, legal and filing fees and are being amortized over the life of the debt. Amortization of the deferred financing costs using the effective interest method was $1.1 million, $0.6 million (as adjusted) and $0.4 million (as adjusted) for the years ended December 31, 2009, 2008 and 2007, respectively, and were included in interest expense. As of December 31, 2009 and 2008, financing costs, net of accumulated amortization were approximately $3.0 million and $4.0 million, respectively.
Revolving Credit Facility
Under our Revolving Credit Facility with Silicon Valley Bank, we may borrow up to the lesser of $25.0 million or 80% of eligible accounts receivable, plus the lesser of 25% of net cash or $10.0 million. As of December 31, 2009, we had $19 million available under the Revolving Credit Facility, of which we borrowed $13 million. All amounts borrowed under the Revolving Credit Facility were repaid in January 2010. All outstanding amounts under the Revolving Credit Facility bear interest at a variable rate equal to the lender’s prime rate plus a margin of 0.25%. In no event shall the interest rate on outstanding borrowings be less than 4.25%, which is payable on a monthly basis. The Revolving Credit Facility also contains customary covenants regarding operations of our business and financial covenants relating to ratios of current assets to current liabilities and is collateralized by all of our assets. An event of default under the Revolving Credit Facility will occur if, among other things, (i) we are delinquent in making payments of principal or interest on the Revolving Credit Facility; (ii) we fail to cure a breach of a covenant or term of the Revolving Credit Facility; (iii) we make a representation or warranty under the Revolving Credit Facility that is materially inaccurate; (iv) we are unable to pay our debts as they become due, certain bankruptcy proceedings are commenced or certain orders are granted against us, or we otherwise become insolvent; (v) an acceleration event occurs under certain types of other indebtedness outstanding from time to time. If an event of default occurs, the indebtedness to Silicon Valley Bank could be accelerated, such that it becomes immediately due and payable. As of December 31, 2009, we were in compliance with all of the covenants under the Revolving Credit Facility. Unless repaid earlier, all amounts owing under the Revolving Credit Facility will become due and payable on December 30, 2010.
Convertible Notes
We issued $40 million in convertible notes in June 2006 which provided, among other things, that if there is a change in control of the Company or certain events of default, as defined, during the term of the convertible notes the holders of the notes will be entitled to receive a cash payment equal to 115% of the principal amount of the notes, plus accrued but unpaid interest, or a formula based on our stock price on certain specified dates. Since the settlement was indexed to our stock, the settlement feature was a derivative that required bifurcation from the host debt instrument and recorded at fair value at each quarter end. We calculated the fair value of the derivative liability by subtracting the value of the convertible notes without the embedded derivative from the value of the convertible notes with the embedded derivative, taking into account the valuation our stock price, the conversion price, the time to maturity of the convertible notes, and the probability of a change in control or certain events of default, as defined on the date of measurement. Changes in fair value of the derivative were recorded as interest expense and the difference between the face value of the convertible notes and the amount remaining after the bifurcation at inception of the notes was recorded as a discount amortized into interest expense over the term of the notes, which was five years.
On September 29, 2008, we repaid all of the outstanding principal and interest under the notes with borrowings under the Facility Agreement. The value of the derivative associated with the convertible notes on the extinguishment date was $0.4 million and was written off as a reduction to loss on extinguishment of debt concurrently with the payoff of the $40 million convertible notes.
Facility Agreement
On September 26, 2008, we entered into a facility agreement, or Facility Agreement, with certain institutional accredited investors, collectively known as Lenders, pursuant to which the Lenders agreed to loan to us up to $65 million, subject to the terms and conditions set forth in the Facility Agreement. At the closing, we initially borrowed $40 million under the Facility Agreement and on October 29, 2008, we borrowed the remaining $25 million. In connection with the financing, we paid Deerfield Management Company, L.P., an affiliate of the lead Lender, a one-time transaction fee of $1.625 million.
Outstanding amounts under the Facility Agreement accrue interest at a rate of 6.5% per annum, payable quarterly in arrears. We are required to repay the Lenders 33% of any principal amount outstanding under the Facility Agreement on each of September 26, 2011 and 2012, and 34% of such principal amount outstanding on September 26, 2013.
F-10
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Additionally, outstanding amounts under the Facility Agreement may become immediately due and payable upon (i) an “event of default,” as defined in the Facility Agreement, in which case the Lenders would have the right to require us to re-pay 100% of the principal amount of the loan, plus any accrued and unpaid interest thereon, or (ii) the consummation of certain change of control transactions, in which case the Lenders would have the right to require us to re-pay 110% of the outstanding principal amount of the loan, plus any accrued and unpaid interest thereon. An event of default under the Facility Agreement will occur if, among other things, (i) we fail to make payment when due; (ii) we fail to comply in any material respect with any covenant of the Facility Agreement, and such failure is not cured; (iii) any representation or warranty made by us in any transaction document was incorrect, false, or misleading in any material respect as of the date it was made; (iv) we are generally unable to pay our debts as they become due or a bankruptcy or similar proceeding has commenced by or against us; and (v) cash and cash equivalents on the last day of each calendar quarter are less than $10 million.
Because the consummation of certain change in control transactions results in a premium of the outstanding principal, the premium put feature is a derivative that is required to be bifurcated from the host debt instrument and recorded at fair value at each quarter end. The value of the derivative at December 31, 2009 was $0.3 million and will be marked-to-market and adjusted quarterly through interest expense.
On December 31, 2009 we had total indebtedness under the Facility Agreement of $65 million, which excludes unamortized discounts of ($7.9) million and the value of the derivative of $0.3 million.
In 2008, we issued to the Lenders warrants to purchase an aggregate of 15 million shares of our common stock at an exercise price of $1.41 per share. If we issue or sell shares of our common stock (other than certain “excluded shares,” as such term is defined in the Facility Agreement), we will issue concurrently therewith additional warrants to purchase such number of shares of common stock as will entitle the Lenders to maintain the same beneficial ownership in the Company after the issuance as they had prior to such issuance, as adjusted on a pro rata basis for repayments of the outstanding principal amount under the loan, with such warrants being issued at an exercise price equal to the greater of $1.41 per share and the closing price of the common stock on the date immediately prior to the issuance.
During 2008, the warrants qualified for classification as equity and their then fair market value of $10.7 million was recorded as additional paid-in capital and discount on debt, to be amortized over the term of the Facility Agreement, or five years.
As required by the Derivatives and Hedging Topic of the FASB Accounting Standards Codification, which provided additional requirements to determine if the warrants were indexed to the Company’s stock, we were required to reclassify our warrants from equity to a liability, effective January 1, 2009. Specifically, because of the anti-dilutive provisions in the warrant agreement, where additional warrants might be issued should we issue additional equity, with such additional warrants being issued at a price equal to the fair value of the common stock being issued, but not less than $1.41, we were required to reclassify the warrants to a liability.
As a result, on January 1, 2009, the warrants were reclassified from equity to a liability and recorded at their estimated fair value. The cumulative effect was a reduction to additional paid-in capital of $10.7 million to reclassify the value of warrants from equity to a liability and a decrease in accumulated deficit of $3.7 million to reflect the change in the value of the warrants between their issuance date and January 1, 2009.
Additionally, the warrants will be marked to market and adjusted quarterly. We recorded a non-cash valuation adjustment of $52.1 million, of $1.57 per share in our financial results for 2009. The change in the valuation of the warrants was primarily driven by an increase in our stock price plus an increase in related volatility.
Commitments and Contingencies
We are subject to routine claims and litigation incidental to our business. In the opinion of management, the resolution of such claims is not expected to have a material adverse effect on our operating results or financial position.
Comprehensive Loss
Accounting Standards Codification 220 “Comprehensive Income”, or ASC 220, requires reporting and displaying comprehensive income (loss) and its components, which, includes net loss and unrealized gains and losses on investments and foreign currency translation gains and losses. Total comprehensive loss for the years ended December 31, 2009 and 2008 was $57.7 million and $34.7 million (as adjusted), respectively. The accumulated balance of unrealized gains (losses) on investments and the accumulated balance of foreign currency translation adjustments are disclosed as separate components of stockholders’ deficit.
F-11
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
As of December 31, 2009 and 2008, accumulated foreign currency translation adjustments were zero and ($25,000), respectively.
Income Taxes
We account for income taxes under the provision of Accounting Standards Codification 740, “Income Taxes”, or ASC 740. As of December 31, 2009 and 2008, there were no unrecognized tax benefits included in the balance sheet that would, if recognized, affect the effective tax rate. Our practice is to recognize interest and/or penalties related to income tax matters in income tax expense. We had no accrual for interest or penalties on our consolidated balance sheets at December 31, 2009 and 2008, respectively and have not recognized interest and/or penalties in the consolidated statement of operations for the year ended December 31, 2009. We are subject to taxation in the United States and various state jurisdictions.
Supply Concentration Risks
Some materials used in our products are currently obtained from a single source. We have a supply agreement with Senju for bepotastine besilate, which is the active pharmaceutical ingredient in Bepreve. Currently, Senju is our sole source for bepotastine besilate. The active ingredient for Xibrom is also supplied to us under an exclusive agreement from a sole supplier. We also have supply agreements with Bausch & Lomb to manufacture commercial quantities of Istalol, Xibrom and Bepreve. Currently, Bausch & Lomb is our sole source for Istalol, Xibrom and Bepreve.
Biozyme Laboratories, Ltd. has been our only source for highly purified ovine hyaluronidase, which is the active ingredient in Vitrase. Our supply agreement with Biozyme ended in August 2007. We are developing and are awaiting qualification from the FDA for an onsite hyaluronidase manufacturing facility. We anticipate receiving qualification of the manufacturing facility by the FDA during the first half of 2010. We entered into a supply agreement with Alliance Medical Products to manufacture commercial quantities of Vitrase. Currently, Alliance Medical Products is our sole source for Vitrase.
Revenue Recognition
Product Revenue. We recognize revenues from product sales when there is persuasive evidence that an arrangement exists, when title has passed, the price is fixed or determinable, and we are reasonably assured of collecting the resulting receivable. We recognize product revenues net of estimated allowances for discounts, product returns, wholesaler fees, rebates and chargebacks. If actual future payments for allowances for discounts, product returns, wholesaler fees, rebates and chargebacks exceed the estimates we made at the time of sale, our financial position, results of operations and cash flows would be negatively impacted.
We establish allowances for estimated rebates, chargebacks and product returns based on numerous qualitative and quantitative factors, including:
| • | the number of and specific contractual terms of agreements with customers; |
| • | estimated level of units in the distribution channel; |
| • | historical rebates, chargebacks and returns of products; |
| • | direct communication with customers; |
| • | anticipated introduction of competitive products or generics; |
| • | anticipated pricing strategy changes by us and/or our competitors; |
| • | analysis of prescription data gathered by a third-party prescription data provider; |
| • | the impact of wholesaler distribution agreements; |
| • | the impact of changes in state and federal regulations; and |
| • | the estimated remaining shelf life of products. |
In our analyses, we utilize on hand unit data purchased from the major wholesalers, as well as prescription data purchased from a third-party data provider, to develop estimates of historical unit channel pull-through. We utilize an internal analysis to compare historical net product shipments to both estimated historical prescriptions written and historical returns. Based on that analysis, we develop an estimate of the quantity of product which may be subject to various discounts, product returns, rebates, chargebacks and wholesaler fees.
We record estimated allowances for rebates, chargebacks, product returns and other discounts, such as wholesaler fees, in the same period when revenue is recognized. The objective of recording the allowances for such deductions at the time of sale is to provide a reasonable estimate of the aggregate amount of credit to our direct customers or payments to our indirect
F-12
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
customers. Customers typically process their claim for allowances such as early pay discounts promptly, usually within the established payment terms. We monitor actual credit memos issued to our customers and compare such actual amounts to the estimated provisions, in the aggregate, for each allowance category to assess the reasonableness of the various reserves at each balance sheet date. Differences between our estimated allowances and actual credits issued have not been significant, and are accounted for in the current period as a change in estimate in accordance with generally accepted accounting principles.
In general, we are obligated to accept from our customers the return of pharmaceutical products that have reached their expiration date. We authorize returns for damaged products and, on a limited basis, exchanges for expiring and expired products in accordance with our return goods policy and procedures, and have established reserves for such amounts at the time of sale. We typically refund the agreed proportion of the sales price by the issuance of a credit, rather than cash refund or exchanges for inventory, and the returned product is destroyed. With the launch of each of our products, we recorded a sales return allowance, which is larger for stocking orders than subsequent re-orders. To date, actual product returns have not exceeded our estimated allowances for returns. Although we believe that our estimates and assumptions are reasonable as of the date when made, actual results may differ significantly from these estimates. Our financial position, results of operations and cash flows may be materially and negatively impacted if actual returns exceed our estimated allowances for returns.
We identify product returns by their manufacturing lot number. Because we manufacture in bulk, lot sizes can be large and, as a result, sales of any individual lot may occur over several periods. As a result, we are unable to specify if actual returns or credits relate to a sale that occurred in the current period or a prior period, and therefore, we cannot specify how much of the allowance recorded relates to sales made in prior periods. Since there have been no material differences between estimates recorded and actual credits issued, we believe our systems and procedures are adequate for managing our business.
Allowances for product returns were $5.5 and $3.2 million as of December 31, 2009 and 2008, respectively. These allowances reflect an estimate of the our liability for products that may be returned by the original purchaser in accordance with our stated return policy, which allows customers to return products within six months of their respective expiration dates and for a period up to twelve months after such products have reached their respective expiration dates. We estimate our liability for product returns at each reporting period based on the estimated units in the channel and the other factors discussed above. Our exposure for product returns has increased as a result of increased sales of our existing products and our additional historical sales data upon which to analyze these allowances. Accordingly, reductions to revenue and corresponding increases to allowance accounts have likewise increased.
As a percentage of gross product revenues, the allowance for product returns was 3.7%, 3.7% and 3.3% for the years ended December 31, 2009, 2008 and 2007, respectively. We typically apply a higher allowance for product returns on stocking orders in connection with an initial launch.
We also periodically offer promotional discounts to our existing customer base. These discounts are calculated as a percentage of the current published list price. Accordingly, the discounts are recorded as a reduction of revenue in the period that the program is offered. In addition to promotional discounts, which are recorded at the time that we implement a price increase, we generally offer our existing customer base an opportunity to purchase a limited quantity of products at the previous list price. Shipments resulting from these programs generally are not in excess of ordinary levels and therefore, we recognize the related revenue upon receipt by the customer and include the sale in estimating our various product-related allowances. In the event we determine that these sales represent purchases of inventory in excess of ordinary levels for a given wholesaler, the potential impact on product returns exposure would be specifically evaluated and reflected as a reduction to revenue at the time of such sale.
Allowances for estimated rebates, chargebacks and other discounts such as wholesaler fees were $6.4 million and $2.9 million as of December 31, 2009 and 2008, respectively. These allowances reflect an estimate of our liability for items such as rebates due to various governmental organizations under the Medicare/Medicaid regulations, rebates due to managed-care organizations under specific contracts, chargebacks due to various organizations purchasing certain of our products through federal contracts and/or group purchasing agreements and fees charged by certain wholesalers under distribution agreements. We estimate our liability for rebates, chargebacks and other discounts such as wholesaler fees at each reporting period based on a combination of quantitative and qualitative assumptions listed above.
As a percentage of gross product revenues, the allowance for rebates, chargebacks and other discounts such as wholesaler fees was 14.7%, 9.8% and 7.5% for the years ended December 31, 2009, 2008 and 2007, respectively. The increase is due primarily to the increase in the number of managed care contracts, federal contracts and wholesaler distribution agreements entered into during 2009 and 2008.
License Revenue. Amounts received for product and technology license fees under multiple-element arrangements are deferred and recognized over the period of such services or performance if such arrangements require on-going services or performance. Amounts received for milestones are recognized upon achievement of the milestone, unless we have ongoing
F-13
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
performance obligations. Any amounts received prior to satisfying our revenue recognition criteria will be recorded as deferred income in the accompanying consolidated balance sheets. During the year ended December 31, 2009, we recognized $3.1 million of previously deferred income primarily related to the termination of our Supply Agreement with Otsuka.
License Fees and Research and Development Costs
Expenditures relating to research and development are expensed in the period incurred. Research and development expenses to date have consisted primarily of costs associated with the clinical trials of our product candidates, compensation and other expenses for research and development personnel, costs for consultants and contract research, costs related to development of commercial scale manufacturing capabilities for our products Xibrom, Istalol, Bepreve and Vitrase and in-process research and development costs related to the acquisition of late-stage development compounds.
We generally classify and separate research and development expenditures into amounts related to clinical development costs, regulatory costs, pharmaceutical development costs, manufacturing development costs and medical affairs costs.
We have expensed amounts paid to acquire licenses, as the ultimate recoverability of the amounts paid was uncertain and the technology had no alternative future use when acquired. Future acquisitions of licenses will be charged to expense or capitalized based upon our assessment regarding the ultimate recoverability of the amounts paid and the potential for alternative future use.
Stock-based Compensation
We recognize compensation expense for all stock-based awards made to employees and directors. The fair value of stock-based awards is estimated at grant date using an option pricing model and the portion that is ultimately expected to vest is recognized as compensation cost over the requisite service period.
Since stock-based compensation is recognized only for those awards that are ultimately expected to vest, we have applied an estimated forfeiture rate to unvested awards for the purpose of calculating compensation cost. These estimates will be revised, if necessary, in future periods if actual forfeitures differ from estimates. Changes in forfeiture estimates impact compensation cost in the period in which the change in estimate occurs.
We use the Black-Scholes option-pricing model to estimate the fair value of stock-based awards. The determination of fair value using the Black-Scholes option-pricing model is affected by our stock price as well as assumptions regarding a number of complex and subjective variables, including expected stock price volatility, risk-free interest rate, expected dividends and projected employee stock option exercise behaviors. We estimate the expected term based on the contractual term of the awards and employees’ exercise and expected post-vesting termination behavior.
At December 31, 2009, there was $5.2 million of total unrecognized compensation cost related to non-vested stock options, which is expected to be recognized over a remaining weighted average vesting period of approximately 2.4 years.
Our stock-based compensation plans are discussed further in Note 4.
Net Loss Per Share
Basic net loss per common share is computed by dividing the net loss for the period by the weighted average number of common shares outstanding during the period. Diluted net loss per share is computed by dividing the net loss for the period by the weighted-average number of common and common equivalent shares, such as stock options, warrants and convertible notes, outstanding during the period. Diluted earnings for common stockholders per common share considers the impact of potentially dilutive securities except in periods in which there is a loss because the inclusion of the potential common shares would have an anti-dilutive effect. Diluted EPS excludes the impact of potential common shares related to our stock options and warrants, in periods in which the options exercise or conversion price is greater than the average market price of our common stock during the period.
Common shares issued for nominal consideration, if any, would be included in the per share calculations as if they were outstanding for all periods presented. We have further determined that the 15 million warrants issued in conjunction with our Facility Agreement in 2008 represented participating securities. However, because we operate at a net loss, and losses are not allocated to the warrant holders, the two class method does not affect our calculation of earnings per share.
F-14
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The following table sets forth the computation of net loss (numerator) and shares (denominator) for loss per share (in thousands):
| Years Ended December 31, | ||||||||||||
| 2009 | 2008 (as adjusted) |
2007 (as adjusted) |
||||||||||
| Numerator: |
||||||||||||
| Net loss |
$ | (57,754 | ) | $ | (34,667 | ) | $ | (41,817 | ) | |||
| Denominator: |
||||||||||||
| Weighted average shares outstanding used for basic loss per share |
33,228 | 33,028 | 29,621 | |||||||||
| Common stock equivalents |
— | — | — | |||||||||
| Weighted average shares outstanding used for diluted loss per share |
33,228 | 33,028 | 29,621 | |||||||||
Potentially dilutive securities, which are not included in our loss per share, are summarized below (in thousands):
| Years Ended December 31, | ||||||
| 2009 | 2008 | 2007 | ||||
| Common stock options |
7,051 | 6,175 | 5,208 | |||
| Warrants |
15,000 | 15,000 | — | |||
| Convertible notes issuance |
— | — | 5,242 | |||
| Total dilutive securities |
22,051 | 21,175 | 10,450 | |||
Equity Financing
In June 2007, we entered into definitive agreements with institutional accredited investors with respect to the private placement of 5.25 million shares of our common stock at a purchase price of $7.00 per share, resulting in gross proceeds of approximately $36.75 million before payment of placement agent fees and offering expenses.
Executive Employment Agreements
We have agreements with each of our officers which provides that any unvested stock options and restricted shares then held by such officer will become fully vested and, with respect to stock options, immediately exercisable, in the event of a change in control of the Company and, in certain instances, if within twenty-four months following such change in control such officer’s employment is terminated by the Company without cause or such officer resigns for good reason within sixty days of the event forming the basis for such good reason termination.
Segment Reporting
We currently operate in only one segment.
Changes in Accounting Principles
Effective January 1, 2009, we changed the classification of our warrants from equity to debt as required by the Derivatives and Hedging Topic of the FASB Accounting Standards Codification. The cumulative effect was a reduction to additional paid-in capital of $10.7 million to reclassify the value of warrants from equity to a liability and a decrease in accumulated deficit of $3.7 million to reflect the change in the value of the warrants between their issuance date and January 1, 2009. The warrants will be carried at estimated fair value and adjusted quarterly through earnings.
Effective January 1, 2009, we have accounted for the liability and equity components of our convertible notes in a manner that reflects our non-convertible debt borrowing rate when interest cost is recognized as required by the Debt with Conversion and Other Options Topic of the FASB Accounting Standards Codification. Additionally, debt issuance costs are required to be allocated in proportion to the allocation of the liability and equity components and accounted for as debt issuance costs and equity issuance costs, respectively. This change required retrospective application and, accordingly, the prior periods’ financial statements included herein have been adjusted.
We determined that the fair value of the convertible notes at issuance in 2006, using a non-convertible debt borrowing rate of 21%, was approximately $28.6 million, and designated the residual value of approximately $11.4 million as the equity component, recorded as a debt discount. Additionally, we allocated approximately $2.0 million of the $2.8 million original convertible notes issuance cost as debt issuance cost and the remaining $0.8 million as equity issuance cost. Because the convertible notes were extinguished in September 2008, the balances of the liability and equity components were zero for each of the periods ended December 31, 2009 and 2008, respectively.
F-15
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The remaining debt discount was amortized to interest expense over the expected life of the convertible notes using the effective interest method, applying a 21% effective interest rate. The convertible notes were issued June 22, 2006 and were due June 22, 2011.
Interest expense related to the convertible notes was recognized as follows (in thousands):
| Years Ended December 31, | |||||||||
| 2009 | 2008 (as adjusted) |
2007 (as adjusted) | |||||||
| Interest expense — stated coupon rate |
$ | — | $ | 2,367 | $ | 3,200 | |||
| Interest expense — amortization of debt discount |
— | 2,842 | 3,789 | ||||||
| Total interest expense — convertible notes |
$ | — | $ | 5,209 | $ | 6,989 | |||
Effect of Change in Accounting Principle to Consolidated Balance Sheet
The following table summarizes the effect of the change in accounting principle related to our convertible notes on the consolidated balance sheet as of December 31, 2008 (in thousands):
| December 31, 2008 | ||||||||||||
| As Previously Reported |
As Adjusted | Effect of Change |
||||||||||
| Assets: |
||||||||||||
| Cash and cash equivalents |
$ | 48,316 | $ | 48,316 | $ | — | ||||||
| Short-term investments |
4,700 | 4,700 | — | |||||||||
| Accounts receivable, net |
15,242 | 15,242 | — | |||||||||
| Inventory, net |
2,289 | 2,289 | — | |||||||||
| Other current assets |
2,150 | 2,150 | — | |||||||||
| Total current assets |
72,697 | 72,697 | — | |||||||||
| Property and equipment, net |
5,728 | 5,728 | — | |||||||||
| Deferred financing costs, net |
4,028 | 4,028 | — | |||||||||
| Deposits and other assets |
207 | 207 | — | |||||||||
| Total assets |
$ | 82,660 | $ | 82,660 | $ | — | ||||||
| Liabilities: |
||||||||||||
| Accounts payable |
$ | 5,386 | $ | 5,386 | $ | — | ||||||
| Accrued compensation and related expenses |
3,945 | 3,945 | — | |||||||||
| Revolving Credit Facility |
15,000 | 15,000 | — | |||||||||
| Current portion of long-term liabilities |
310 | 310 | — | |||||||||
| Current portion of obligations under capital leases |
117 | 117 | — | |||||||||
| Allowance for rebates and chargebacks |
2,074 | 2,074 | — | |||||||||
| Allowance for product returns |
3,241 | 3,241 | — | |||||||||
| Royalties payable |
5,867 | 5,867 | — | |||||||||
| Other accrued expenses |
5,257 | 5,257 | — | |||||||||
| Total current liabilities |
41,197 | 41,197 | — | |||||||||
| Deferred rent |
213 | 213 | — | |||||||||
| Deferred income |
3,055 | 3,055 | — | |||||||||
| Obligations under capital leases |
187 | 187 | — | |||||||||
| Long-term liabilities |
50 | 50 | — | |||||||||
| Facility Agreement |
55,157 | 55,157 | — | |||||||||
| Total liabilities |
99,859 | 99,859 | — | |||||||||
| Stockholders’ deficit: |
||||||||||||
| Common stock |
33 | 33 | — | |||||||||
| Additional paid-in capital |
316,650 | 326,036 | 9,386 | |||||||||
| Accumulated other comprehensive loss |
(25 | ) | (25 | ) | — | |||||||
| Accumulated deficit |
(333,857 | ) | (343,243 | ) | (9,386 | ) | ||||||
| Total stockholders’ deficit |
(17,199 | ) | (17,199 | ) | — | |||||||
| Total liabilities and stockholders’ deficit |
$ | 82,660 | $ | 82,660 | $ | — | ||||||
F-16
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Effect of Change in Accounting Principle to Consolidated Statements of Operations
The following table summarizes the effect of change in accounting principle related to our convertible notes on the consolidated statements of operations for the years ended December 31, 2008 and 2007 (in thousands):
| Year Ended December 31, 2008 |
||||||||||||
| As Previously Reported |
As Adjusted | Effect of Change |
||||||||||
| Total revenues |
$ | 83,076 | $ | 83,076 | $ | — | ||||||
| Cost of products sold |
21,947 | 21,947 | — | |||||||||
| Costs and expenses |
85,939 | 85,939 | — | |||||||||
| Loss from operations |
(24,810 | ) | (24,810 | ) | — | |||||||
| Other (expense) income: |
||||||||||||
| Interest income |
714 | 714 | — | |||||||||
| Interest expense |
(5,403 | ) | (8,100 | ) | (2,697 | ) | ||||||
| Loss on extinguishment of debt |
(1,201 | ) | (2,497 | ) | (1,296 | ) | ||||||
| Gain on derivative valuation |
26 | 26 | — | |||||||||
| Total other expense, net |
(5,864 | ) | (9,857 | ) | (3,993 | ) | ||||||
| Net loss |
$ | (30,674 | ) | $ | (34,667 | ) | (3,993 | ) | ||||
| Net loss per common share, basic and diluted |
$ | (0.93 | ) | $ | (1.05 | ) | $ | (0.12 | ) | |||
| Shares used in computing net loss per common share, basic and diluted |
33,028 | 33,028 | — | |||||||||
| Year Ended December 31, 2007 |
||||||||||||
| As Previously Reported |
As Adjusted | Effect of Change |
||||||||||
| Total revenues |
$ | 58,867 | $ | 58,867 | $ | — | ||||||
| Cost of products sold |
15,864 | 15,864 | — | |||||||||
| Costs and expenses |
79,095 | 79,095 | — | |||||||||
| Loss from operations |
(36,092 | ) | (36,092 | ) | — | |||||||
| Other (expense) income: |
||||||||||||
| Interest income |
2,141 | 2,141 | — | |||||||||
| Interest expense |
(4,073 | ) | (7,669 | ) | (3,596 | ) | ||||||
| Loss on derivative valuation |
(197 | ) | (197 | ) | — | |||||||
| Total other expense, net |
(2,129 | ) | (5,725 | ) | (3,596 | ) | ||||||
| Net loss |
$ | (38,221 | ) | $ | (41,817 | ) | (3,596 | ) | ||||
| Net loss per common share, basic and diluted |
$ | (1.29 | ) | $ | (1.41 | ) | $ | (0.12 | ) | |||
| Shares used in computing net loss per common share, basic and diluted |
29,621 | 29,621 | — | |||||||||
F-17
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Effect of Change in Accounting Principle to Consolidated Statements of Cash Flows
The following table summarizes the effect of change in accounting principle related to our convertible notes on the consolidated statements of cash flows for the years ended December 31, 2008 and 2007 (in thousands):
| Year Ended December 31, 2008 |
||||||||||||
| As Previously Reported |
As Adjusted | Effect of Change |
||||||||||
| Operating activities |
||||||||||||
| Net loss |
$ | (30,674 | ) | $ | (34,667 | ) | $ | (3,993 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Stock-based compensation costs |
3,978 | 3,978 | — | |||||||||
| Amortization of deferred financing costs |
687 | 569 | (118 | ) | ||||||||
| Amortization of discount on convertible notes |
27 | 2,842 | 2,815 | |||||||||
| Amortization of discount on Facility Agreement |
544 | 544 | — | |||||||||
| Loss on extinguishment of debt |
1,201 | 2,497 | 1,296 | |||||||||
| Change in value of derivative related to convertible notes |
42 | 42 | — | |||||||||
| Change in value of derivative related to Facility Agreement |
354 | 354 | — | |||||||||
| Depreciation and amortization |
1,046 | 1,046 | — | |||||||||
| Changes in operating assets and liabilities |
||||||||||||
| Accounts receivable, net |
(4,081 | ) | (4,081 | ) | — | |||||||
| Inventory, net |
5 | 5 | — | |||||||||
| Other current assets |
(28 | ) | (28 | ) | — | |||||||
| Accounts payable |
1,725 | 1,725 | — | |||||||||
| Accrued compensation and related expenses |
249 | 249 | — | |||||||||
| Other accrued expenses |
2,964 | 2,964 | — | |||||||||
| Other liabilities |
(241 | ) | (241 | ) | — | |||||||
| Deferred rent |
(17 | ) | (17 | ) | — | |||||||
| Deferred income |
(278 | ) | (278 | ) | — | |||||||
| Net cash used in operating activities |
(22,497 | ) | (22,497 | ) | — | |||||||
| Investing activities |
||||||||||||
| Net cash provided by investing activities |
16,984 | 16,984 | — | |||||||||
| Financing activities |
||||||||||||
| Net cash provided by financing activities |
28,329 | 28,329 | — | |||||||||
| Increase in cash and cash equivalents |
22,816 | 22,816 | ||||||||||
| Cash and cash equivalents at beginning of year |
25,500 | 25,500 | — | |||||||||
| Cash and cash equivalents at end of year |
$ | 48,316 | $ | 48,316 | $ | — | ||||||
F-18
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
| Year Ended December 31, 2007 |
||||||||||||
| As Previously Reported |
As Adjusted | Effect of Change |
||||||||||
| Operating activities |
||||||||||||
| Net loss |
$ | (38,221 | ) | $ | (41,817 | ) | $ | (3,596 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Stock-based compensation costs |
3,488 | 3,488 | — | |||||||||
| Amortization of deferred financing costs |
558 | 401 | (157 | ) | ||||||||
| Amortization of discount on convertible notes |
56 | 3,809 | 3,753 | |||||||||
| Change in value of derivative related to convertible notes |
197 | 197 | — | |||||||||
| Depreciation and amortization |
725 | 725 | — | |||||||||
| Changes in operating assets and liabilities |
||||||||||||
| Accounts receivable, net |
(4,822 | ) | (4,822 | ) | — | |||||||
| Inventory, net |
(820 | ) | (820 | ) | — | |||||||
| Other current assets |
(450 | ) | (450 | ) | — | |||||||
| Accounts payable |
1,443 | 1,443 | — | |||||||||
| Accrued compensation and related expenses |
1,385 | 1,385 | — | |||||||||
| Other accrued expenses |
5,012 | 5,012 | — | |||||||||
| Other liabilities |
(285 | ) | (285 | ) | — | |||||||
| Deferred rent |
14 | 14 | — | |||||||||
| Deferred income |
(278 | ) | (278 | ) | — | |||||||
| Net cash used in operating activities |
(31,998 | ) | (31,998 | ) | — | |||||||
| Investing activities |
||||||||||||
| Net cash provided by investing activities |
1,378 | 1,378 | — | |||||||||
| Financing activities |
||||||||||||
| Net cash provided by financing activities |
38,884 | 38,884 | — | |||||||||
| Increase in cash and cash equivalents |
8,264 | 8,264 | ||||||||||
| Cash and cash equivalents at beginning of year |
17,236 | 17,236 | — | |||||||||
| Cash and cash equivalents at end of year |
$ | 25,500 | $ | 25,500 | $ | — | ||||||
New Accounting Pronouncements
In May 2009, the FASB issued Accounting Standards Codification 855-10, “Subsequent Events”, or ASC 855-10, which establishes general standards for accounting and disclosure of events that occur after the balance sheet date but before the financial statements are issued or are available to be issued. The pronouncement requires the disclosure of the date through which an entity has evaluated subsequent events and the basis for that date, whether that date represents the date the financial statements were issued or were available to be issued. We evaluated subsequent events through the issuance date of the financial statements, which did not have a material impact on our consolidated financial statements.
In June 2009, the FASB issued Accounting Standards Codification 105-10, “Accounting Standards Codification and the Hierarchy of Generally Accepted Accounting Principles”, or ASC 105-10. ASC 105-10 became the source of authoritative U.S. Generally Accepted Accounting Principles, or GAAP, recognized by the FASB to be applied by non-government entities. It also modified the GAAP hierarchy to include only two levels of GAAP; authoritative and non-authoritative. We adopted ASC 105-10 for the reporting in 2009. The adoption did not have a material impact on our consolidated financial statements.
In August 2009, the FASB issued Accounting Standards Update No. 2009-05, Measuring Liabilities at Fair Value, or ASU 2009-05, which amends ASC 820 to provide clarification of a circumstance in which a quoted price in an active market for an identical liability is not available. A reporting entity is required to measure fair value using one or more of the following methods: 1) a valuation technique that uses a) the quoted price of the identical liability when traded as an asset or b) quoted prices for similar liabilities (or similar liabilities when traded as assets) and/or 2) a valuation technique that is consistent with the principles of ASC 820. ASU 2009-05 also clarifies that when estimating the fair value of a liability, a reporting entity is not required to adjust to include inputs relating to the existence of transfer restrictions on that liability. The adoption did not have a material impact on our consolidated financial statements.
F-19
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
2. Balance Sheet Details
Inventory
Inventories are stated at the lower of cost (first-in, first-to-expire) or market. Inventories at December 31, 2009 and 2008 consist of the following (in thousands):
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Raw materials |
$ | 2,626 | $ | 1,367 | ||||
| Finished goods |
3,818 | 1,534 | ||||||
| 6,444 | 2,901 | |||||||
| Less reserve for excess and obsolescence |
(896 | ) | (612 | ) | ||||
| $ | 5,548 | $ | 2,289 | |||||
Property and Equipment
Equipment and leasehold improvements and related accumulated depreciation and amortization are as follows (in thousands):
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Equipment |
$ | 7,893 | $ | 6,617 | ||||
| Furniture and fixtures |
944 | 944 | ||||||
| Equipment under capital leases |
538 | 419 | ||||||
| Leasehold improvements |
1,276 | 1,274 | ||||||
| 10,651 | 9,254 | |||||||
| Less accumulated depreciation and amortization |
(4,535 | ) | (3,526 | ) | ||||
| $ | 6,116 | $ | 5,728 | |||||
Total depreciation and amortization expense amounted to $1.0 million, $1.0 million and $0.7 million for the years ended December 31, 2009, 2008 and 2007, respectively.
3. Short-Term Investments and Fair Value Measurements
Short-term Investments
We had no short-term investments as of December 31, 2009. We classified our short-term investments as “trading” at December 31, 2008 (consisting solely of auction rate securities) and recorded such assets at the estimated fair value. Unrealized gains and losses on auction rate securities were included in earnings. The basis for computing realized gains or losses was by specific identification.
In February 2008, auctions began to fail for the auction rate securities we held and each auction since then had failed. Consequently, the investments were not liquid at December 31, 2008. In November 2008, we accepted an offer (a “Right”), from UBS, our investment provider for the auction rate securities, entitling us to sell at par value auction rate securities originally purchased from UBS at anytime during a two-year period from January 2, 2009 through January 4, 2011. In accepting the Right, we granted UBS the authority to sell or auction the auction rate securities at par at any time up until the expiration date of the offer and release UBS from any claims relating to the marketing and sale of auction rate securities. In lieu of our acceptance of the Right, auction rate securities continued to accrue and pay interest as determined by the auction process or the terms specified in the prospectus of the auction rate security if the auction process failed.
As of December 31, 2008, we recorded $0.7 million as the fair value of the put option asset, classified as short-term investment on the Consolidated Balance Sheet as of December 31, 2008, with a corresponding credit to interest expense in the Consolidated Statement of Operations for the year ended December 31, 2008. During the year ended December 31, 2008, we recognized $0.7 million on the gain of the put right from UBS fully offset by the loss on the auction rate securities. As of December 31, 2009 and 2008, there were no unrealized gains and losses. On January 2, 2009, we exercised our Right and received $4.7 million in par value plus accrued interest.
The following is a summary of the investments held at December 31, 2008, excluding put option from UBS (in thousands):
| Gross Amortized Cost |
Net Unrealized Gains/ (Losses) |
Estimated Fair Value | |||||||
| Auction Rate securities |
$ | 3,971 | $ | — | $ | 3,971 | |||
| $ | 3,971 | $ | — | $ | 3,971 | ||||
F-20
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Fair Value Measurements
We account for fair value measurements under FASB Accounting Standard Codification 820 “Fair Value Measurements and Disclosures”, or ASC 820, which defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants at the measurement date. ASC 820 establishes a three-level fair value hierarchy that prioritizes the inputs used to measure fair value. This hierarchy requires entities to maximize the use of observable inputs and minimize the use of unobservable inputs. The three levels of inputs used to measure fair value are as follows:
| • | Level 1 — Quoted prices in active markets for identical assets or liabilities. |
| • | Level 2 — Observable inputs other than quoted prices included in Level 1, such as quoted prices for similar assets and liabilities in active markets; quoted prices for identical or similar assets and liabilities in markets that are not active; or other inputs that are observable or can be corroborated by observable market data. |
| • | Level 3 — Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. This includes certain pricing models, discounted cash flow methodologies and similar techniques that use significant unobservable inputs. |
We have segregated all assets and liabilities measured at fair value on a recurring basis (at least annually) into the most appropriate level within the fair value hierarchy based on the inputs used to determine the fair value at the measurement date in the table below. As of December 31, 2009, all of our assets and liabilities are valued using Level 1 inputs except for a derivative and warrants related to our Facility Agreement.
Assets and liabilities measured at fair value on a recurring basis are summarized below (in thousands):
| Fair Value Measurements at December 31, 2009 Using: |
Total Carrying Value at December 31, 2009 |
||||||||||||||
| Level 1 | Level 2 | Level 3 | |||||||||||||
| Cash and cash equivalents |
$ | 53,702 | $ | — | $ | — | $ | 53,702 | |||||||
| Derivatives (Facility Agreement) |
— | — | (299 | ) | (299 | ) | |||||||||
| Warrants |
— | (58,663 | ) | — | (58,663 | ) | |||||||||
| Fair Value Measurements Using Significant Unobservable Inputs (Level 3) Auction Rate Securities |
Fair Value Measurements Using Significant Unobservable Inputs (Level 3) Put Option |
|||||||
| Beginning balance at December 31, 2008 |
$ | 3,971 | $ | 729 | ||||
| Total gains or losses (realized or unrealized) |
||||||||
| Included in earnings |
— | — | ||||||
| Included in other comprehensive income |
— | — | ||||||
| Purchases, issuances, and settlements |
(3,971 | ) | (729 | ) | ||||
| Transfers in and/or out of Level 3 |
— | — | ||||||
| Ending balance at December 31, 2009 |
$ | — | $ | — | ||||
| Fair Value Measurements Using Significant Unobservable Inputs (Level 3) Derivative |
Fair Value Measurements Using Observable Inputs Other Than Level 1 (Level 2) Warrants |
|||||||
| Beginning balance at December 31, 2008 |
$ | (1,476 | ) | $ | — | |||
| Reclassification and adjustment pursuant to ASC 815-40(1) |
— | (6,597 | ) | |||||
| Total gains or losses (realized or unrealized) |
||||||||
| Included in earnings |
1,177 | (52,066 | ) | |||||
| Included in other comprehensive income |
— | — | ||||||
| Purchases, issuances, and settlements |
— | — | ||||||
| Transfers in and/or of Level 3 |
— | — | ||||||
| Ending balance at December 31, 2009 |
$ | (299 | ) | $ | (58,663 | ) | ||
| (1) | Reflects reclassification of warrants and mark to market adjustment of $3.7 million as of January 1, 2009 (see Note 1). |
F-21
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
4. Stockholders’ Equity
Common and Preferred Stock
In June 2006, we issued an aggregate of $40.0 million in principal amount of our convertible notes, bearing 8% interest per annum payable quarterly in cash in arrears. The notes were scheduled to mature in June 2011 and were convertible into shares of our common stock at a conversion price of $7.63 per share, subject to anti-dilution adjustments. On September 26, 2008, we repaid all of the outstanding principal amount and interest on the convertible notes.
In June 2007, we entered into definitive agreements with institutional accredited investors with respect to the private placement of 5,250,000 shares of our common stock at a purchase price of $7.00 per share, resulting in gross proceeds of approximately $36.7 million before payment of placement agent fees and offering expenses.
At December 31, 2009 and 2008, we had 5,000,000 shares of preferred stock authorized at a $0.001 par value and no shares were issued and outstanding.
Common Stock Warrants
During September and October 2008, we issued a total of 15 million warrants at an exercise price of $1.41 per share in conjunction with our borrowing of $65 million under our Facility Agreement. If we issue or sell shares of our common stock (other than certain “excluded shares,” as such term is defined in the Facility Agreement), we will issue concurrently therewith additional warrants to purchase such number of shares of common stock as will entitle the Lenders to maintain the same beneficial ownership in the Company after the issuance as they had prior to such issuance, as adjusted on a pro rata basis for repayments of the outstanding principal amount under the loan, with such warrants being issued at an exercise price equal to the greater of $1.41 per share and the closing price of the common stock on the date immediately prior to the issuance.
The warrants expire on September 26, 2014 and contain certain limitations that prevent the holder from acquiring shares upon exercise of a warrant that would result in the number of shares beneficially owned by it to exceed 9.98% of the total number of shares of our common stock then issued and outstanding.
In addition, upon certain change of control transactions, or upon certain “events of default” (as defined in the warrants), the holder has the right to net exercise the warrants for an amount of shares our common stock equal to the Black-Scholes value of the shares issuable under the warrants divided by 95% of the closing price of the common stock on the day immediately prior to the consummation of such change of control or event of default, as applicable. In certain circumstances where a warrant or portion of a warrant is not net exercised in connection with a change of control or event of default, the holder will be paid an amount in cash equal to the Black-Scholes value of such portion of the warrant not treated as a net exercise.
Employee Stock Purchase Plan
In April 2000, our Board of Directors adopted the Employee Stock Purchase Plan, or 2000 ESPP, which initially provided for the issuance of a maximum of 20,000 shares of common stock. The 2000 ESPP provided for annual increases in the number of shares available for issuance under the plan on the first day of each year, beginning in 2001, equal to the lesser of 20,000 shares, 1.5% of the outstanding shares of common stock on the first day of the year, or a lesser amount as the Board of Directors may determine.
Under the 2000 ESPP, eligible employees could have had up to 15% of their earnings withheld, subject to certain maximums, to be used to purchase shares of our common stock every January and July. The price of the common stock purchased under the 2000 ESPP was equal to 85% of the lower of the fair value of the common stock on the commencement date of the offering period or the specified purchase date. During 2009, 2008 and 2007, 20,208 shares, 22,788 shares, 18,336 shares, respectively, had been issued to participants, with an additional 9,557 shares issuable at December 31, 2009.
On December 7, 2009, our stockholders approved the 2009 Employee Stock Purchase Plan, or 2009 ESPP. The 2009 ESPP replaces our existing 2000 ESPP, which will expire pursuant to its terms in April 2010. No further purchase rights will be granted under our existing 2000 ESPP.
Initially, an aggregate of 3,000,000 shares will be reserved for issuance under the 2009 ESPP. In addition, on each January 1, beginning on January 1, 2011, the number of shares reserved will be increased by the lesser of (i) 1% of the Company’s outstanding common stock or (ii) an amount determined by the Compensation Committee, or any other administrator of the 2009 ESPP. However, in no event will the number of shares reserved exceed the lesser of 10% of our outstanding common stock or 5,000,000 shares.
F-22
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Every employee of the Company who customarily works more than 20 hours per week for more than five months per calendar year will be eligible to participate in offerings made under the 2009 ESPP, subject to certain limitations. Shares of common stock will generally be offered for purchase through a series of six-month offering periods. The initial offering period will commence on January 1, 2010 and end on June 30, 2010, with subsequent offering periods commencing on six-month intervals thereafter beginning on July 31, 2010. The purchase price for the common stock will be the lower of 85% of the fair market value of the common stock on the first day of an offering period or 85% of the fair market value of the common stock on the last day of the offering period.
The 2009 ESPP will terminate on October 18, 2019, unless earlier terminated in accordance with the terms and provisions of the 2009 ESPP.
Stock Compensation Plan
Stock Options
We have outstanding options to purchase shares of our common stock under individual option agreements, our 1993 Stock Plan and our 2000 Stock Plan. All of the outstanding options granted under the individual option agreements, the 1993 Stock Plan and the 2000 Stock Plan will remain outstanding and subject to the provisions of the applicable agreement and plan until they are either exercised or expire in accordance with their respective terms. No options were issued under the 1993 Stock Plan after the adoption of the 2000 Stock Plan as shares of common stock available for future issuance under the 1993 Stock Plan were assumed under the 2000 Stock Plan. No options were awarded under the 2000 Stock Plan after the adoption of the 2004 Stock Plan. Any shares available for future issuance under the 2000 Stock Plan have been included in the shares of common stock authorized for issuance under the 2004 Performance Incentive Plan, as amended, or 2004 Stock Plan.
The 2004 Stock Plan provides for the grant of stock options, restricted stock awards, and performance shares to qualified employees, officers, directors, consultants and other service providers. The 2004 Stock Plan originally authorized us to grant options and/or rights to purchase up to an aggregate of 2,053,107 shares of common stock. In October 2005, the options available for issuance under the 2004 Stock Plan were increased by 1,000,000 shares to 3,053,107, of which up to 300,000 shares may be issued in connection with restricted stock awards or performance share awards. In October 2006, the options available for issuance under the 2004 Stock Plan was increased by 3,100,000 shares to 6,153,107, of which up to 700,000 shares may be issued in connection with restricted stock awards or performance share awards. In December 2009, the options available for issuance under the 2004 Stock Plan was increased by 6,000,000 shares to an aggregate of 12,153,107 shares, of which up to 1,450,000 shares may be issued in connection with restricted stock awards or performance share awards.
As of December 31, 2009, a total of 6,917,871 shares of common stock were reserved for issuance under the 2004 Stock Plan. A summary of our stock option activity and related information during 2009 follows:
| Number of Shares |
Weighted- Average Exercise Price |
Remaining Contractual Life |
Aggregate Intrinsic Value | ||||||||
| Outstanding at December 31, 2008 |
6,174,913 | $ | 6.18 | ||||||||
| Granted |
1,270,688 | $ | 2.68 | ||||||||
| Exercised |
(52,425 | ) | $ | 3.16 | |||||||
| Canceled |
(341,713 | ) | $ | 6.20 | |||||||
| Outstanding at December 31, 2009 |
7,051,463 | $ | 5.57 | 6.27 | $ | 5,256,931 | |||||
| Options vested and expected to vest at December 31, 2009 |
6,706,922 | $ | 5.66 | 6.16 | $ | 4,805,829 | |||||
| Exercisable at December 31, 2009 |
4,889,971 | $ | 6.40 | 5.19 | $ | 2,349,889 | |||||
F-23
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The aggregate intrinsic value of options exercised during the years ended December 31, 2009, 2008 and 2007 was $272,976, $8,063 and $270,798, respectively.
ESPP activity during 2009 was as follows:
| Number of Shares |
Weighted- Average Purchase Price | |||||
| Available at December 31, 2008 |
202 | |||||
| New shares added to ESPP |
20,000 | |||||
| Purchases |
(19,700 | ) | $ | 2.14 | ||
| Available at December 31, 2009 |
502 | |||||
Restricted stock activity during 2009 was as follows:
| Number of Shares |
Weighted- Average Fair Value | |||||
| Outstanding at December 31, 2008 |
302,279 | $ | 4.94 | |||
| Granted |
159,434 | $ | 1.55 | |||
| Vested |
(102,750 | ) | $ | 4.81 | ||
| Forfeited |
(11,307 | ) | $ | 2.54 | ||
| Outstanding at December 31, 2009 |
347,656 | $ | 3.50 | |||
The weighted average fair value of equity instruments granted during 2009, 2008 and 2007 was as follows:
| Weighted Average Fair Value | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Stock options |
$ | 2.68 | $ | 3.31 | $ | 7.15 | |||
| ESPP Purchases |
2.14 | 1.21 | 5.06 | ||||||
| Restricted Stock |
1.55 | 3.40 | 6.95 | ||||||
At December 31, 2009, there was $5.2 million of total unrecognized compensation cost, related to non-vested stock options, which is expected to be recognized over a remaining weighted average vesting period of 2.4 years.
We use the Black-Scholes option-pricing model to estimate the fair value of stock-based awards. The determination of fair value using the Black-Scholes option-pricing model is affected by our stock price as well as assumptions regarding a number of complex and subjective variables, including expected stock price volatility, risk-free interest rate, expected dividends and projected employee stock option exercise behaviors. We estimate the expected term based on the contractual term of the awards and employees’ exercise and expected post-vesting termination behavior.
The total number of stock option awards expected to vest is adjusted by estimated forfeiture rates. The weighted average assumptions used for the years ended December 31, 2009, 2008 and 2007 and the resulting estimates of weighted-average fair value per share of options granted and for stock purchases under the ESPP during those periods are as follows:
| Years Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Interest rate |
2.0 | % | 2.5 | % | 4.3 | % | |||
| Volatility |
91.0 | % | 74.0 | % | 70.0 | % | |||
| Expected life |
7 years | 7 years | 6 years | ||||||
| Expected dividend yield |
0 | % | 0 | % | 0 | % | |||
Stock-based compensation costs are as follows (in millions):
| Years Ended December 31, | |||||||||
| 2009 | 2008 | 2007 | |||||||
| Selling, general and administrative |
$ | 2.6 | $ | 3.0 | $ | 2.5 | |||
| Research and development |
1.2 | 1.0 | 1.0 | ||||||
| Stock-based compensation costs |
$ | 3.8 | $ | 4.0 | $ | 3.5 | |||
F-24
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Restricted Stock Awards
During the years ended December 31, 2009, 2008 and 2007, we granted a total of 159,434, 170,817 and 154,553 shares of restricted common stock, respectively, to employees under the 2004 Stock Plan. Restrictions on these shares will expire and related charges are being amortized as earned over the vesting period of four years.
The amount of unearned compensation recorded is based on the market value of the shares on the date of issuance. Expenses related to the vesting of restricted stock were $0.6 million, $0.6 million and $0.3 million for the years ended December 31, 2009, 2008 and 2007, respectively. As of December 31, 2009, there was approximately $0.7 million of unamortized compensation cost related to restricted stock awards, which is expected to be recognized ratably over the vesting period of four years.
5. Commitments and Contingencies
Legal
In April 2008, we received subpoenas from the office of the U.S. Attorney for the Western District of New York requesting information regarding the marketing activities related to Xibrom. From April 2008 through December 31, 2009, we incurred approximately $2.8 million in legal fees associated with this matter since receipt of the subpoenas and expect to incur significant expenses in the future. In addition, if the government chooses to engage in civil litigation or initiate a criminal prosecution against us as a result of its review of the requested documents and other evidence, we may have to incur significant amounts to defend such action or pay or incur substantial fines or penalties, either of which could significantly deplete our cash resources. The case is in its preliminary stages and thus the likelihood of an unfavorable outcome and/or the amount/range of loss, if any, cannot be reasonably estimated.
We are involved in other legal proceedings incidental to our business from time to time. Except as described immediately above, we do not believe that pending actions or proceedings, either individually or in the aggregate, will have a material adverse effect on our financial condition, results of operations or cash flows, and adequate provision has been made for the resolution of such actions and proceedings.
TRICARE Retail Pharmacy Program
The Department of Defense, or DoD’s, TRICARE Retail Pharmacy program pursuant to section 703 of the National Defense Authorization Act of 2008, became effective on May 26, 2009. This regulation would require manufacturers to pay rebates to the DoD on products distributed to TRICARE beneficiaries through retail pharmacies retroactive to January 28, 2008. The regulation requires that pharmaceuticals paid for by the DoD through the TRICARE Retail Pharmacy program be subject to the Federal Ceiling Price program, which would require manufacturers to provide DoD with a refund on pharmaceuticals utilized through the TRICARE Retail Pharmacy program. We have requested a waiver of the retroactive rebate for TRICARE Retail Pharmacy utilization for the period from January 28, 2008 to May 26, 2009. In addition, the regulation is currently the subject of litigation, and it is our belief that the retroactive application of the regulation is contrary to established case law. We have determined that payment of the retroactive rebate created by the regulation is neither reasonably estimable nor probable as of December 31, 2009.
Manufacturing Facility
Ovine hyaluronidase, the active pharmaceutical ingredient used in Vitrase, is processed in several stages to produce a highly purified raw material for formulation. We had a supply agreement with Biozyme Laboratories, Ltd., or Biozyme, for ovine hyaluronidase. Our supply agreement with Biozyme ended in August 2007. We are developing and are awaiting qualification from the FDA for an onsite hyaluronidase manufacturing facility. We anticipate receiving qualification of the manufacturing facility by the FDA during the first half of 2010. Our success in validating the manufacturing site for production of ovine hyaluronidase cannot be assured.
Equipment Financing
During 2006, we entered into a credit arrangement under which we borrowed $1.2 million to finance the purchase of capital equipment. The outstanding amounts under this arrangement are due and payable ratably over three years from the date of each loan schedule. As of December 31, 2009 and 2008, $14,000 and $271,000, respectively, were outstanding under this arrangement.
F-25
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Debt and Lease Commitments
We lease our corporate and laboratory facilities and certain equipment under various operating leases. As of December 31, 2009, we have made approximately $0.2 million in cash deposits related to operating leases. Provisions of the facilities lease provide for abatement of rent during certain periods and escalating rent payments during the term. Rent expense is recognized on a straight-line basis over the term of the lease. Accordingly, rent expense recognized in excess of rent paid is reflected as deferred rent. Additionally, we are required to pay taxes, insurance and maintenance expenses related to the building. Rent expense on the facilities and equipment during 2009, 2008 and 2007 was $0.8 million, $0.8 million and $0.7 million, respectively.
Future annual minimum payments under operating leases are as follows (in thousands):
| Years Ending December 31: |
|||
| 2010 |
$ | 1,057 | |
| 2011 |
199 | ||
| 2012 |
165 | ||
| 2013 |
171 | ||
| 2014 |
179 | ||
| Thereafter |
231 | ||
| $ | 2,002 | ||
Scheduled maturities of capital leases, debt, amounts borrowed under the Revolving Credit Facility and Facility Agreement as of December 31, 2009, are as follows (in thousands):
| Years Ending December 31: |
||||
| 2010 |
$ | 13,156 | ||
| 2011 |
21,533 | |||
| 2012 |
21,470 | |||
| 2013 |
22,110 | |||
| 2014 |
— | |||
| Thereafter |
— | |||
| $ | 78,269 | |||
| Unamortized discount on Facility Agreement |
(7,089 | ) | ||
| Embedded derivative on Facility Agreement |
299 | |||
| $ | 71,479 | |||
Milestones
In addition to the above, we are committed to make potential future milestone payments to third parties as part of our in-licensing and development programs. Milestone payments under these agreements generally become due and payable only upon achievement of certain development, regulatory and/or commercial milestones. Because the achievement of these milestones is neither probable nor reasonably estimable, such contingencies have not been recorded on our consolidated balance sheet. As of December 31, 2009, the maximum potential future milestone payments to third parties are $27.0 million.
6. Income Taxes
We account for income taxes under the Income Tax Topic of the FASB Accounting Standards Codification. As of December 31, 2009 and 2008, there is no unrecognized tax benefits included in the balance sheet that would, if recognized, affect the effective tax rate.
Our practice is to recognize interest and/or penalties related to income tax matters in income tax expense. We had no accrual for interest or penalties on our consolidated balance sheets at December 31, 2009 and 2008, respectively and have not recognized interest and/or penalties in the consolidated statement of operations for the year ended December 31, 2009.
We are subject to taxation in the United States and various state jurisdictions. Our tax years for 1994 and forward are subject to examination by the United States and state tax authorities.
At December 31, 2009, we had net deferred tax assets of $46.3 million. Due to uncertainties surrounding our ability to generate future taxable income to realize these assets, a full valuation has been established to offset the net deferred tax asset. Additionally, the future utilization of our net operating loss and research and development credit carry forwards to offset
F-26
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
future taxable income may be subject to an annual limitation, pursuant to Internal Revenue Code Sections 382 and 383, as a result of ownership changes that may have occurred previously or that could occur in the future. We have not performed a Section 382 analysis to determine the limitation of the net operating loss and research and development credit carry forwards. Until this analysis has been performed, we have removed the deferred tax assets for net operating losses of $65.8 million and research and development credits of $15.9 million generated through 2009 from its deferred tax asset schedule, and have recorded a corresponding decrease to its valuation allowance. When this analysis is finalized, we plan to update our unrecognized tax benefits. Due to the existence of the valuation allowance, future changes in our unrecognized tax benefits will not impact our effective tax rate.
At December 31, 2009, we had Federal and state income tax net operating loss carry forwards of approximately $178.9 million and $106.3 million, respectively. Federal tax loss carryforwards continue to expire in 2009 unless previously utilized. California tax loss carry forwards begin to expire in 2012, unless previously utilized. In addition, we have Federal and California research and development tax credit carry forwards of $9.7 million and $6.2 million, respectively. The Federal research and development credit carry forwards will begin to expire in 2010 unless previously utilized. The California research and development credit carry forwards carry forward indefinitely.
Our deferred tax asset was $46.3 million for 2009 as compared to $46.2 million for 2008, or an increase of $0.1 million. Significant components of our deferred tax assets as of December 31, 2009 and 2008 are listed below. A valuation allowance of $46.3 million and $46.2 million at December 31, 2009 and 2008, respectively, has been recognized to offset the net deferred tax assets as realization of such assets is uncertain. Our valuation allowance changed by $0.1 million, $8.6 million, and ($63.0) million (primarily as a result of the aforementioned removal of the deferred tax assets for net operating losses) for the years ended December 31, 2009, 2008 and 2007, respectively. A summary of the components of our deferred taxes follows (in thousands):
| December 31, | ||||||||
| 2009 | 2008 | |||||||
| Deferred tax asset: |
||||||||
| Capitalized research and development |
$ | 39,465 | $ | 39,082 | ||||
| Stock based compensation |
3,785 | 3,497 | ||||||
| Deferred income |
— | 1,215 | ||||||
| Other, net |
3,071 | 2,379 | ||||||
| Total deferred tax asset |
46,321 | 46,173 | ||||||
| Valuation allowance for deferred tax assets |
(46,321 | ) | (46,173 | ) | ||||
| $ | — | $ | — | |||||
A portion of the net operating loss carry forwards as of December 31, 2009 include amounts related to stock option deductions. Any excess tax benefits from share-based compensation are only realized when income taxes payable is reduced, with the corresponding credit posted to Additional Paid-in Capital.
7. Employee Benefit Plan
We have a 401(k) Savings Plan covering substantially all employees that have been employed for one month and meet certain age requirements. Employees may contribute up to 92% of their compensation per year (subject to a maximum limit by federal tax law). In 2009, we provided matching contributions equal to 25% of the first 6% of contributed salary. Employer contributions were $0.2 million, $0.2 million and $0.1 million for the years ended December 31, 2009, 2008 and 2007, respectively.
8. Senju Agreements
In May 2002, we acquired substantially all of the assets of AcSentient, which included exclusive U.S. marketing rights for Istalol and Xibrom. Istalol and Xibrom were originally licensed by AcSentient from Senju. The full rights and obligations of AcSentient under the Senju license agreements were assigned to us as a part of the acquisition agreement between us and AcSentient, with such transfer approved by Senju.
In November 2004, we entered into another license agreement with Senju under which Senju granted us exclusive U.S. marketing rights to Senju’s ecabet sodium product for the treatment of dry eye syndrome.
In 2006, we entered into three additional license agreements with Senju under which Senju granted us exclusive North American rights for Bepreve for the treatment of ocular allergy, latanoprost to treat glaucoma and iganidipine to enhance blood flow to the optic nerve.
F-27
Table of Contents
ISTA PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
In December 2009, we entered into agreements amending the terms of the Xibrom license agreement by expanding our territory rights to include Canada and Mexico. This agreement is effective January 1, 2010.
Generally, under the terms of our agreements with Senju, we are responsible for all costs associated with developing the licensed products in ophthalmology for the United States and, with respect to bepotastine, prostaglandins and iganidipine, North America, including clinical trials, regulatory filings, manufacturing, and, if the product is approved, marketing and sales activities.
We have paid to Senju non-refundable milestone payments of $4 million relating to the development process and regulatory approval of both Istalol and Xibrom; and will be required to pay royalties on product sales. We will also be required to pay to Senju non-refundable milestone payments of up to $3 million, some of which has been paid, if all such milestones, relating to the development process and regulatory approval of ecabet sodium are accomplished; and royalties on future product sales. We have paid to Senju non-refundable milestone payments of $4 million relating to the development process and regulatory approval of Bepreve; and will be required to pay royalties on product sales. According to our iganidipine agreement with Senju, we will be required to pay Senju non-refundable milestone payments of approximately $8 million, some of which has been paid, if all such milestones, relating to the development process and regulatory approval of iganidipine are accomplished; and royalties on future product sales. In addition, under the terms of our prostaglandins agreement with Senju, we will be required to pay Senju non-refundable milestone payments of approximately $8 million, some of which has been paid, if all such milestones, relating to the development process and regulatory approval of prostaglandins are accomplished; and royalties on future product sales.
9. Mitsubishi Tanabe Agreement
During September 2007, we licensed exclusive North American rights to nasal dosage forms of bepotastine, an investigational product for the treatment of allergy symptoms, from Mitsubishi Tanabe Pharma Corporation (formerly Tanabe Seiyaku Co., Ltd.). Under the terms of the license agreement with Mitsubishi Tanabe we paid an upfront payment to Mitsubishi Tanabe of $2.0 million, and will make additional payments based on achievement of development and approval milestones, and royalties on future product sales. We are responsible for all costs associated with developing nasal bepotastine in North America, including clinical trials, FDA filings, manufacturing, and, if the product is approved, marketing and sales activities. We also obtained the right to develop other nasal bepotastine products, including a fixed combination with a steroid, and a future right to negotiate for a North American license to oral dosage forms of bepotastine for allergy treatment.
10. Quarterly Results of Operations (unaudited)
The following table sets forth a summary of our unaudited quarterly operating results for each of the last eight quarters in the period ended December 31, 2009. This data has been derived from our unaudited consolidated interim financial statements which, in our opinion, have been prepared on substantially the same basis as the audited financial statements contained elsewhere in this report and include all normal recurring adjustments necessary for a fair presentation of the financial information for the periods presented. These unaudited quarterly results should be read in conjunction with our financial statements and notes thereto included elsewhere in this report. The operating results in any quarter are not necessarily indicative of the results that may be expected for any future period (in thousands except earnings per share).
| Quarter Ended | ||||||||||||||||||||||||||||||||
| Dec. 31, 2009 |
Sept. 30, 2009 |
June 30, 2009 |
Mar. 31, 2009 |
Dec. 31, 2008 (as adjusted) |
Sept. 30, 2008 (as adjusted) |
June 30, 2008 (as adjusted) |
Mar. 31, 2008 (as adjusted) |
|||||||||||||||||||||||||
| (Unaudited) | ||||||||||||||||||||||||||||||||
| Revenues: |
||||||||||||||||||||||||||||||||
| Product sales, net |
$ | 34,284 | $ | 29,080 | $ | 23,884 | $ | 20,345 | $ | 28,049 | $ | 21,604 | $ | 17,691 | $ | 15,454 | ||||||||||||||||
| License revenue |
— | 2,917 | 69 | 69 | 70 | 69 | 70 | 69 | ||||||||||||||||||||||||
| Total revenues |
34,284 | 31,997 | 23,953 | 20,414 | 28,119 | 21,673 | 17,761 | 15,523 | ||||||||||||||||||||||||
| Cost of products sold |
8,631 | 7,304 | 6,214 | 5,129 | 7,186 | 5,806 | 4,766 | 4,189 | ||||||||||||||||||||||||
| Gross profit margin |
25,653 | 24,693 | 17,739 | 15,285 | 20,933 | 15,867 | 12,995 | 11,334 | ||||||||||||||||||||||||
| Costs and expenses: |
||||||||||||||||||||||||||||||||
| Research and development |
5,261 | 6,257 | 6,644 | 6,742 | 7,347 | 7,766 | 7,472 | 9,815 | ||||||||||||||||||||||||
| Selling, general and administrative |
17,503 | 13,187 | 12,708 | 12,979 | 13,389 | 13,198 | 13,302 | 13,650 | ||||||||||||||||||||||||
| Total costs and expenses |
22,764 | 19,444 | 19,352 | 19,721 | 20,736 | 20,964 | 20,774 | 23,465 | ||||||||||||||||||||||||
| Income (loss) from operations |
2,889 | 5,249 | (1,613 | ) | (4,436 | ) | 197 | (5,097 | ) | (7,779 | ) | (12,131 | ) | |||||||||||||||||||
| Other expense, net |
(3,468 | ) | (6,166 | ) | (35,033 | ) | (15,176 | ) | (2,085 | ) | (4,471 | ) | (1,578 | ) | (1,723 | ) | ||||||||||||||||
| Net loss |
$ | (579 | ) | $ | (917 | ) | $ | (36,646 | ) | $ | (19,612 | ) | $ | (1,888 | ) | $ | (9,568 | ) | $ | (9,357 | ) | $ | (13,854 | ) | ||||||||
| Net loss per common share, basic and diluted |
$ | (0.02 | ) | $ | (0.03 | ) | $ | (1.10 | ) | $ | (0.59 | ) | $ | (0.06 | ) | $ | (0.29 | ) | $ | (0.28 | ) | $ | (0.42 | ) | ||||||||
F-28
Table of Contents
SCHEDULE II — VALUATION AND QUALIFYING ACCOUNTS
| Description |
Balance at beginning of year |
Additions | Deductions | Balance at end of year |
|||||||||||
| (in thousands) | |||||||||||||||
| Allowance for Rebates and Chargebacks: |
|||||||||||||||
| Year ended December 31, 2009 |
$ | (2,074 | ) | $ | (13,298 | ) | $ | 10,593 | $ | (4,779 | ) | ||||
| Year ended December 31, 2008 |
(1,501 | ) | (5,612 | ) | 5,039 | (2,074 | ) | ||||||||
| Year ended December 31, 2007 |
(892 | ) | (3,193 | ) | 2,584 | (1,501 | ) | ||||||||
| Allowance for Product Returns |
|||||||||||||||
| Year ended December 31, 2009 |
$ | (3,241 | ) | $ | (4,927 | ) | $ | 2,659 | $ | (5,509 | ) | ||||
| Year ended December 31, 2008 |
(1,738 | ) | (3,549 | ) | 2,046 | (3,241 | ) | ||||||||
| Year ended December 31, 2007 |
(760 | ) | (2,200 | ) | 1,222 | (1,738 | ) | ||||||||
| Allowance for Doubtful Accounts |
|||||||||||||||
| Year ended December 31, 2009 |
$ | (134 | ) | $ | 13 | $ | 27 | $ | (94 | ) | |||||
| Year ended December 31, 2008 |
(187 | ) | (258 | ) | 311 | (134 | ) | ||||||||
| Year ended December 31, 2007 |
(121 | ) | (89 | ) | 23 | (187 | ) | ||||||||
Table of Contents
EXHIBIT INDEX
| Exhibit |
Description | |
| 2.1 | Asset Purchase and Sale Agreement dated May 3, 2002, by and between the Registrant and AcSentient, Inc. (Incorporated by reference to Exhibit 2.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on May 6, 2002). | |
| 3.1 | Restated Certificate of Incorporation of Registrant (Incorporated by reference to Exhibit 3.1 of the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2002, filed with the Commission on March 7, 2003). | |
| 3.2 | Certificate of Correction to Restated Certificate of Incorporation of Registrant (Incorporated by reference to Exhibit 3.2 of the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2002, filed with the Commission on March 7, 2003). | |
| 3.3 | Second Certificate of Correction to Restated Certificate of Incorporation of Registrant (Incorporated by reference to Exhibit 3.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on August 31, 2005). | |
| 3.4 | Amended and Restated Bylaws of Registrant (Incorporated by reference to Exhibit 3.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on October 31, 2006). | |
| 4.1 | Specimen common stock certificate (Incorporated by reference to Exhibit 4.1 of the Registrant’s Registration Statement on Form S-1/A (File No. 333-34120) filed with the Commission on August 7, 2000). | |
| 4.2 | Preferred Stock Rights Agreement dated as of December 31, 2001, by and between the Registrant and Mellon Investor Services LLC, as rights agent (Incorporated by reference to Exhibit 4.2 of the Registrant’s Registration Statement on Form 8-A (File No. 000-31255) filed with the Commission on January 22, 2002). | |
| 4.3 | First Amendment to the Preferred Stock Rights Agreement dated as of November 18, 2002, by and between the Registrant and Mellon Investor Services LLC, as rights agent (Incorporated by reference to Exhibit 4.2 of the Registrant’s Registration Statement on Form 8-A12G/A (File No. 000-31255) filed with the Commission on November 19, 2002). | |
| 4.4 | Second Amendment to the Preferred Stock Rights Agreement, dated June 23, 2006, by and between the Registrant and U.S. Stock Transfer Corporation (Incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K filed with the Securities and Exchange Commission on June 28, 2006). | |
| 10.1 | 1993 Stock Plan and forms of agreements thereunder (Incorporated by reference to Exhibit 10.2 of the Registrant’s Registration Statement on Form S-1 (File No. 333-34120) filed with the Commission on April 5, 2000). (2) | |
| 10.2 | 2000 Stock Plan (Amended and Restated) (Incorporated by reference to Exhibit 10.1 of the Registrant’s Quarterly Report on Form 10-Q for the period ended June 30, 2003, filed with the Commission on August 14, 2003). (2) | |
| 10.3 | Forms of agreements under 2000 Stock Plan (Incorporate by reference to Exhibit 10.3 of the Registrant’s Registration Statement on Form S-1 (File No. 333-34120) filed with the Commission on April 5, 2000). (2) | |
| 10.4 | 2009 Employee Stock Purchase Plan (Incorporated by reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8-K filed with the Commission on December 11, 2009). (2) | |
Table of Contents
| Exhibit Number |
Description | |
| 10.5 | Fourth Amendment and Restatement to the 2004 Performance Incentive Plan (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on December 11, 2009). (2) | |
| 10.6 | Form of Stock Option Agreement under 2004 Performance Incentive Plan (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed with the Commission on August 31, 2005). (2) | |
| 10.7 | Form of Restricted Stock Purchase Agreement under 2004 Performance Incentive Plan (Incorporated by reference to Exhibit 10.3 of the Registrant’s Current Report on Form 8-K filed with the Commission on August 31, 2005). (2) | |
| 10.8 | Form of Indemnification Agreement by and between the Registrant and certain executive officers and directors of Registrant (Incorporated by reference to Exhibit 10.8 of the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2005, filed with the Commission on March 6, 2006). (2) | |
| 10.8.1 | Schedule of Parties to Indemnification Agreement (Incorporated by reference to Exhibit 10.10.1 of the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2007, filed with the Commission on March 7, 2008). | |
| 10.9 | Lease by and between the Registrant and Aetna Life Insurance Company dated September 13, 1996 for leased premises located at 15279 Alton Parkway, Suite 100, Irvine, California (Incorporated by reference to Exhibit 10.9 of the Registrant’s Registration Statement on Form S-1 (File No. 333-34120) filed with the Commission on April 5, 2000). | |
| 10.10 | First Amendment to Lease by and between the Registrant and Alton Plaza Property, Inc. dated June 27, 2001 for leased premises located at 15279 Alton Parkway, Suite 100, Irvine, California (Incorporated by reference to Exhibit 10.30 of the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2001, filed with the Commission on April 1, 2002). | |
| 10.11 | Second Amendment to Lease by and between the Registrant and Alton Plaza Property, Inc. dated February 13, 2002 for leased premises located at 15279 Alton Parkway, Suite 100, Irvine, California (Incorporated by reference to Exhibit 10.31 of the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2001, filed with the Commission on April 1, 2002). | |
| 10.12 | Third Amendment to Lease by and between the Registrant and Alton Plaza Property, Inc. dated August 13, 2004 for leased premises located at 15279 Alton Parkway, Suite 100, Irvine, California (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on August 13, 2004). | |
| 10.13 | Fourth Amendment to Lease by and between the Registrant and Alton Plaza Property, Inc., dated for reference purposes only as of August 31, 2005, for the lease of office space located at 15273 Alton Parkway, Irvine, California (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on September 14, 2005). | |
| 10.14 | Fifth Amendment to Lease by and between the Registrant and Alton Plaza Property, Inc., dated for reference purposes only as of July 25, 2007, for the lease of the office space located at 15285 Alton Parkway, Irvine, California (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on August 21, 2007). | |
| 10.15 | License Agreement dated as of December 13, 2001, by and between Otsuka Pharmaceutical Co., Ltd., and the Registrant (Incorporated by reference to Exhibit 10.21 of the Registrant’s Current Report on Form 8-K filed with the Commission on January 2, 2002). (1) | |
| 10.16 | Executive Employment Agreement dated March 13, 2006, by and between Vicente Anido, Jr., Ph.D. and the Registrant (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on March 15, 2006). (2) | |
| 10.17 | Executive Employment Agreement dated February 10, 2003, by and between Lauren P. Silvernail and the Registrant (Incorporated by reference to Exhibit 10.1 of the Registrant’s Quarterly Report on Form 10-Q for the period ended March 31, 2003, filed with the Commission on May 15, 2003). (2) | |
Table of Contents
| Exhibit |
Description | |
| 10.18 | Form of Executive Employment Agreement by and between the Registrant and certain executive officers of the Registrant, each entered into on March 13, 2006 (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed with the Commission on March 15, 2006). (2) | |
| 10.18.1 | Schedule of Parties to Executive Employment Agreement (Incorporated by reference to Exhibit 10.21.1 of the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2007, filed with the Commission on March 7, 2008). | |
| 10.19 | Stand-Alone Stock Option Agreement dated December 21, 2001, by and between Vicente Anido, Jr., Ph.D. and the Registrant (Incorporated by reference to Exhibit 10.28 of the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2001, filed with the Commission on April 1, 2002). (2) | |
| 10.20 | Change of Control Severance Agreement dated February 20, 2003 by and between Lauren Silvernail and the Registrant (Incorporated by reference to Exhibit 10.17 of the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2005, filed with the Commission on March 6, 2006). (2) | |
| 10.21 | Individual Non-Qualified Stock Option Agreement dated July 1, 2002, by and between Thomas A. Mitro and the Registrant (Incorporated by reference to Exhibit 99.1 of the Registrant’s Registration Statement on Form S-8 (File No. 333-103279) filed with the Commission on February 18, 2003). (2) | |
| 10.22 | Individual Non-Qualified Stock Option Agreement dated August 5, 2002, by and between Kirk McMullin and the Registrant (Incorporated by reference to Exhibit 99.2 of the Registrant’s Registration Statement on Form S-8 (File No. 333-103279) filed with the Commission on February 18, 2003). (2) | |
| 10.23 | Bausch & Lomb Pharmaceuticals, Inc. Contract Manufacturing Supply Agreement dated February 6, 2003, by and between Bausch & Lomb Pharmaceuticals, Inc. and the Registrant (Incorporated by reference to Exhibit 10.37 of the Registrant’s Annual Report on Form 10-K/A for the year ended December 31, 2002, filed with the Commission on June 4, 2003). (1) | |
| 10.24 | Bausch & Lomb Pharmaceuticals, Inc. Contract Manufacturing Supply Agreement dated November 25, 2002, by and between Bausch & Lomb Pharmaceuticals, Inc. and the Registrant (Incorporated by reference to Exhibit 10.38 of the Registrant’s Annual Report on Form 10-K/A for the year ended December 31, 2002, filed with the Commission on June 4, 2003). (1) | |
| 10.25 | Agreement dated April 17, 2002, by and between Senju Pharmaceutical Co., Ltd. and AcSentient, Inc. (Incorporated by reference to Exhibit 10.43 of the Registrant’s Annual Report on Form 10-K/A for the year ended December 31, 2002, filed with the Commission on June 4, 2003). (1) | |
| 10.26 | Amendment to Timolol Agreement dated August 13, 2002, by and between Senju Pharmaceutical Co., Ltd. and the Registrant (Incorporated by reference to Exhibit 10.46 of the Registrant’s Annual Report on Form 10-K/A for the year ended December 31, 2002, filed with the Commission on April 30, 2003). (1) | |
| 10.27 | License Agreement dated March 7, 2002, by and between Senju Pharmaceutical Co., Ltd and AcSentient, Inc. (Incorporated by reference to Exhibit 10.42 of the Registrant’s Annual Report on Form 10-K/A for the year ended December 31, 2002, filed with the Commission on June 4, 2003). (1) | |
| 10.28 | Amendment to Bromfenac License Agreement dated August 13, 2002, by and between Senju Pharmaceutical Co., Ltd and the Registrant (Incorporated by reference to Exhibit 10.45 of the Registrant’s Annual Report on Form 10-K/A for the year ended December 31, 2002, filed with the Commission on April 30, 2003). (1) | |
| 10.29 | Second Amendment to Bromfenac License Agreement dated May 31, 2006, by and between Senju Pharmaceutical Co., Ltd and the Registrant (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on June 2, 2006). | |
| 10.30 | Letter Agreement, dated December 11, 2009, by and between Senju Pharmaceutical Co., Ltd and the Registrant (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on December 17, 2009). | |
| 10.31 | Letter Agreement, dated December 11, 2009, by and between Senju Pharmaceutical Co., Ltd and the Registrant (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed with the Commission on December 17, 2009). (1) | |
| 10.32 | Amended and Restated Supply Agreement dated June 16, 2004, by and between Biozyme Laboratories, Ltd. and the Registrant (Incorporated by reference to Exhibit 99.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on June 21, 2004). | |
| 10.33 | Technology License Agreement dated June 16, 2004, by and between Biozyme Laboratories, Ltd. and the Registrant (Incorporated by reference to Exhibit 99.2 of the Registrant’s Current Report on Form 8-K filed with the Commission on June 21, 2004). | |
| 10.34 | Vitrase® Agreement dated September 27, 2004, by and between the Registrant and Allergan Sales, LLC (Incorporated by reference to Exhibit 10.1 of the Registrant’s Quarterly Report on Form 10-Q for the period ended September 30, 2004, filed with the Commission on November 12, 2004). (1) | |
Table of Contents
| Exhibit |
Description | |
| 10.35 | License Agreement dated November 17, 2004, by and between the Registrant and Senju Pharmaceuticals Co., Ltd. (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K/A filed with the Commission on December 28, 2004). (1) | |
| 10.36 | Supply Agreement dated August 30, 2004, by and between the Registrant and Alliance Medical Products, Inc. (Incorporated by reference to Exhibit 10.45 of the Registrant’s Annual Report on Form 10-K for the year ended December 31, 2004, filed with the Commission on March 15, 2005). (1) | |
| 10.37 | Loan and Security Agreement dated December 22, 2005, by and between the Registrant and Silicon Valley Bank (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on December 22, 2005). | |
| 10.38 | Loan Modification Agreement dated September 26, 2006, by and between the Registrant and Silicon Valley Bank (Incorporated by reference to Exhibit 10.32.1 of the Registrant’s Quarterly Report on Form 10-Q for the period ended September 30, 2006, filed with the Commission on November 9, 2006). | |
| 10.39 | Loan Modification Agreement dated January 30, 2007, by and between the Registrant and Silicon Valley Bank (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on February 2, 2007). | |
| 10.40 | Loan Modification Agreement dated March 12, 2007, by and between the Registrant and Silicon Valley Bank (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on March 14, 2007). | |
| 10.41 | Loan Modification Agreement dated March 26, 2008, by and between the Registrant and Silicon Valley Bank (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on March 31, 2008). | |
| 10.42 | Loan Modification Agreement dated June 30, 2008, by and between the Registrant and Silicon Valley Bank (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on July 3, 2008). | |
| 10.43 | Loan Modification Agreement dated September 26, 2008, by and between the Registrant and Silicon Valley Bank (Incorporated by reference to Exhibit 10.5 of the Registrant’s Current Report on Form 8-K filed with the Commission on September 30, 2008). | |
| 10.44 | Loan Modification Agreement dated December 23, 2008, by and between the Registrant and Silicon Valley Bank (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on December 24, 2008). | |
| 10.45 | Loan Modification Agreement, dated December 16, 2009, by and between the Registrant and Silicon Valley Bank (Incorporated by reference to Exhibit 10.3 of the Registrant’s Current Report on Form 8-K filed with the Commission on December 17, 2009). | |
| 10.46 | Exclusive License Agreement dated June 12, 2006, by and between the Registrant and Senju Pharmaceutical Co., Ltd. (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on June 16, 2006). (1) | |
| 10.47 | Exclusive License Agreement dated June 12, 2006, by and between the Registrant and Senju Pharmaceutical Co., Ltd. (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed with the Commission on June 16, 2006). (1) | |
| 10.48 | Securities Purchase Agreement, dated as of June 21, 2006, by and between the Registrant and the investors named therein (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on June 22, 2006). | |
| 10.49 | Registration Rights Agreement dated as of June 21, 2006, by and between the Registrant and the investors named therein (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed with the Commission on June 22, 2006). | |
| 10.50 | Form of Senior Subordinated Convertible Note (Incorporated by reference to Exhibit 10.3 of the Registrant’s Current Report on Form 8-K filed with the Commission on June 22, 2006). | |
| 10.51 | Exclusive License Agreement dated August 1, 2006, by and between the Registrant and Senju Pharmaceutical Co., Ltd. (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on August 3, 2006). (1) | |
| 10.52 | Form of Purchase Agreement (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on June 27, 2007). | |
Table of Contents
| Exhibit |
Description | |
| 10.53 | Exclusive License Agreement dated September 25, 2007, by and between Registrant and Mitsubishi Tanabe Pharma Corporation (formerly Tanabe Seiyaku Co., Ltd.) Co., Ltd. (Incorporated by reference to Exhibit 10.47 of the Registrant’s Quarterly Report on Form 10-Q for the period ended September 30, 2007, filed with the Commission on November 6, 2007). (1) | |
| 10.54 | Form of Warrant to purchase shares of common stock of Registrant (Incorporated by reference to Exhibit 4.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on September 30, 2008). | |
| 10.55 | Facility Agreement dated September 26, 2008 by and between the Registrant and certain lenders named therein (Incorporated by reference to Exhibit 10.1 of the Registrant’s Current Report on Form 8-K filed with the Commission on September 30, 2008). | |
| 10.56 | Amendment dated September 26, 2008 by and between the Registrant and Highbridge International LLC (Incorporated by reference to Exhibit 10.2 of the Registrant’s Current Report on Form 8-K filed with the Commission on September 30, 2008). | |
| 10.57 | Registration Rights Agreement dated September 26, 2008 by and between the Registrant and certain investors named therein (Incorporated by reference to Exhibit 10.3 of the Registrant’s Current Report on Form 8-K filed with the Commission on September 30, 2008). | |
| 10.58 | Security Agreement dated September 26, 2008 by and between the Registrant and certain secured parties named therein (Incorporated by reference to Exhibit 10.4 of the Registrant’s Current Report on Form 8-K filed with the Commission on September 30, 2008). | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 23.2 | Consent of Independent Registered Public Accounting Firm. | |
| 24.1 | Power of Attorney (included in the signature page). | |
| 31.1 | Certification of Chief Executive Officer Pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934. | |
| 31.2 | Certification of Chief Financial Officer Pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934. | |
| 32.1 | Certification of Chief Executive Officer Pursuant to Rule 13a-14(b)/15d-14(b) of the Securities Exchange Act of 1934 and 18 U.S.C. Section 1350. (3) | |
| 32.2 | Certification of Chief Financial Officer Pursuant to Rule 13a-14(b)/15d-14(b) of the Securities Exchange Act of 1934 and 18 U.S.C. Section 1350. (3) | |
| (1) | Portions of this exhibit are omitted and were filed separately with the Secretary of the Commission pursuant to ISTA’s application requesting confidential treatment under Rule 24b-2 of the Exchange Act of the Securities Exchange Act of 1934. |
| (2) | These exhibits are identified as management contracts or compensatory plans or arrangements of the Registrant pursuant to Item 15(a)(3) of Form 10-K. |
| (3) | Furnished herewith and not “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended. |